
У человека глаукома наследуется как аутосомно рецессивный
Обновлено: 30.06.2024
Появление на свет ребенка с врожденными дефектами развития всегда ошеломляет семью; эта тема - одна из самых тяжелых в акушерстве. Супруги в первый момент испытывают ни с чем не сравнимый психологический шок, который затем переходит в чувство вины, им кажется, что у них уже никогда не будет здорового ребенка.
Следует сразу сказать, что ребенок с врожденными пороками может появиться на свет абсолютно в любой семье - молодой, здоровой, без вредных привычек, с нормально протекающей беременностью. По данным многолетней статистики, во всем мире около 5% детей рождается с врожденными заболеваниями.
Врожденные пороки развития плода можно разделить на две большие группы - наследственно обусловленные (то есть заложенные в генах и хромосомах, передающиеся по наследству) и собственно врожденные (приобретенные в ходе внутриутробного развития). Такое деление довольно условно, так как большинство дефектов развития вызываются сочетанием наследственной предрасположенности и неблагоприятного внешнего воздействия, представляя собой мультифакториальные аномалии.
Проблема врожденных пороков развития плода очень многообразна, изучением этого вопроса занимаются различные специалисты - генетики, неонатологи, эмбриологи, специалисты по дородовой (пренатальной) диагностике. Разобраться в причинах всегда бывает непросто.
Наследственно обусловленные заболевания
В основе наследственно обусловленных заболеваний лежат мутации. Благодаря современным леденящим кровь триллерам, слово это вызывает сейчас у многих почти суеверный ужас. На самом деле латинское слово mutatio означает "изменение" - не более того. Мутация - это изменение наследственных свойств организма в результате перестроек в структурах, ответственных за хранение и передачу генетической информации. Заболевания, связанные с патологическими изменениями в хромосомах, обычно так и называют хромосомными заболеваниями. Под собственно наследственными заболеваниями понимают нарушения, обусловленные генными мутациями.
В ядрах соматических (неполовых) клеток содержится по 23 пары хромосом, из которых одна пара - половые хромосомы. У женщин эта пара состоит из двух одинаковых хромосом, условно называемых Х-хромосомами, у мужчин эти хромосомы разные - Х-хромосома и Y-хромосома. Неполовые хромосомы называются аутосомами.
В половых клетках хромосом в два раза меньше - не 23 пары, а 23 штуки.
При оплодотворении ядра яйцеклетки и сперматозоида сливаются, и будущий человечек получает полный набор хромосом, наследуя таким образом и материнские, и отцовские признаки.
Хромосомы состоят из генов. За каждый признак в организме отвечает пара генов - "мамин" и "папин". (Исключение составляет XY-пара половых хромосом у мужчин: не все гены Х-хромосомы имеют "напарников" в Y-хромосоме.) В каждой паре один ген доминирует (доминантный ген), т.е. проявляется обусловленный им вариант признака, другой - "уступает" (рецессивный ген). При неблагоприятном стечении обстоятельств оба гена в паре или один из них могут оказаться носителями патологического признака. В первом случае их "владелец", без сомнения, болен. Если же мы имеем дело лишь с одним "больным" геном, возможны два варианта: (1) за заболевание "отвечает" доминантный ген - тогда его носитель болен; (2) носитель патологического признака - рецессивный ген - тогда человек здоров (точнее, как говорят врачи, фенотипически здоров, т.е. при наличии "больного" гена в генотипе, отсутствуют какие-либо проявления болезни).
Аутосомно-доминантный тип наследования
Носитель патологического признака - доминантный ген, содержащийся в аутосоме (неполовой хромосоме). При этом типе наследования невозможно рождение больного ребенка у здоровых родителей - хотя бы один из родителей страдает от того же заболевания. При этом мальчики и девочки в равной степени подвержены заболеванию. Такие дефекты развития, как правило, бывают негрубыми и после успешной коррекции не препятствуют нормальной жизни.
Аутосомно-рецессивный тип наследования
Носитель патологического признака - рецессивный ген, содержащийся в аутосоме. При аутосомно-рецессивном механизме наследования ситуация выглядит парадоксально - у здоровых родителей вдруг появляется на свет ребенок с дефектами развития, порой тяжелейшими и даже несовместимыми с жизнью. Причина - носительство обоими супругами в скрытом состоянии мутантных рецессивных генов. При этом рождение больного ребенка не обязательно означает, что все следующие дети будут страдать тем же заболеванием. Так же, как и в аутосомно-доминантном типе, мальчики и девочки в равной степени подвержены заболеванию.
Сцепленное с полом рецессивное наследование
Пороки развития, сцепленные с полом, в основном обусловлены рецессивными мутациями в женской половой хромосоме (этот тип наследования называют еще Х-хромосомным). Такой признак всегда передается через мать - носительницу рецессивного "больного" гена (т.е. сама женщина здорова). Практически все пораженные - мужчины (у пораженного гена Х-хромосомы в Y-хромосоме отсутствует "партнер", который мог бы доминировать над ним). Больной мужчина никогда не передает заболевания своим сыновьям (ведь они получают от него "здоровую" Y-, а не мутантную Х-хромосому), однако все его дочери будут носительницами "рокового" гена.
Мы схематически описали типы наследования, чтобы дать читателю общее представление о сути этих механизмов. На самом деле все гораздо сложнее - куда менее однозначно и определенно.
В приведенной ниже таблице перечислены в качестве примера лишь некоторые из совместимых с жизнью наследственных аномалий
Механизм наследования
Лечебно-реабилитационные меры
Аутосомно-рецессивное наследование - возможно рождение ребенка-альбиноса от здоровых родителей. Частота в популяции 1:20 000
Отсутствие нормальной пигментации кожи, волос, радужной оболочки глаза
Эта наследственная аномалия не считается заболеванием в полном смысле этого слова и лечению не подлежит
Сцепленное с полом рецессивное наследование. Болеют главным образом мужчины. Передается от матери сыновьям
Заболевание обусловлено дефицитом некоторых факторов свертывания крови. Проявляется кровоточивостью
Лечение при кровотечении - переливание крови, плазмы; кровоостанавливающие средства общего действия; антигемофильный глобулин; профилактика травм и кровотечений
Сцепленное с полом рецессивное наследование. Наблюдается преимущественно у мужчин. Передается от матери сыновьям
Частичная цветовая слепота. Распространяется чаще всего на красный и зеленый цвета
Расстройство цветового зрения выявляют при помощи специальных таблиц или спектральных приборов. Дальтонизм лечению не подлежит
Хромосомная аномалия: у матери при созревании яйцеклетки под влиянием пока не выясненных причин в 21-й паре хромосом образуется 3 хромосомы вместо 2-х. Частота в популяции - 1:700
Одна из форм врожденного слабоумия. Степень психического недоразвития значительно колеблется. Больные в основном ласковы, добродушны, приветливы
Лечебная педагогика, основанная на склонности больных к подражательности. Обучение во вспомогательных школах, трудотерапия
Аутосомно-доминантное наследование, передается детям от родителей с врожденной формой заболевания
Опущение верхнего века вследствие недоразвития мышцы, поднимающей его
Врожденные мультифакториальные пороки развития
- Ионизирующее излучение (рентгеновские лучи, воздействие радиоактивных изотопов). Кроме прямого действия на генетический аппарат, ионизирующее излучение обладает токсическим эффектом и является причиной многих врожденных аномалий
- Тератогенные инфекции, т.е. инфекционные заболевания, передающиеся от матери плоду
- Медикаменты. Нет лекарств, которые могут быть безоговорочно признаны полностью безопасными, особенно на ранних стадиях беременности. Во время беременности лучше воздержаться от приема лекарств любого типа - за исключением, конечно, случаев, когда это необходимо для спасения жизни или ликвидации серьезной угрозы для здоровья матери или плода
- Алкоголь. Употребление алкоголя приводит к разнообразным врожденным нарушениям, выраженность которых зависит от количества употребляемого алкоголя - особенно на ранних стадиях беременности. Фетальный (т.е. поражающий плод) алкогольный синдром - тяжелейшее врожденное заболевание, порой несовместимое с жизнью.
- Никотин. Курение сигарет во время беременности приводит к отставанию ребенка в физическом развитии
- Воздействие токсических химических веществ
Зачастую, однако, в развитии врожденных пороков играет роль такой фактор, как наследственная предрасположенность: известно, что если у родителей или ближайших родственников наблюдались врожденные пороки развития, то риск родить ребенка со сходными дефектами повышается, то есть речь идет "о семейном накоплении" аномалий развития. Так, у женщины с врожденным пороком сердца шансы родить ребенка с дефектом развития сердечно-сосудистой системы несколько выше, чем у всех остальных женщин. Поэтому принято говорить не столько о просто врожденных, сколько о врожденных мультифакториальных пороках развития. Тем не менее, на большом статистическом материале показано, что повторный риск рождения ребенка с врожденным пороком развития невелик - в среднем 2-4%.
Приведем несколько примеров совместимых с жизнью врожденных мультифакториальных пороков развития
Дефект развития
Выхождение внутренних органов или глубоких тканей из полостей, обычно занимаемых ими, под кожу или в межмышечную клетчатку без нарушения целости покровов
Массаж, в случае его неэффективности - хирургическое лечение
Врожденный вывих и врожденная дисплазия тазобедренного сустава
Врожденная дисплазия тазобедренного сустава - недоразвитие тканей тазобедренного сустава, отсутствие соответствия между суставными поверхностями - состояние, предшествующее вывиху тазобедренного сустава
При дисплазии - применение различных ортезов (приспособлений для отведения бедер) у детей до года. При вывихе - вправление, наложение специальных ортезов в первые месяцы жизни. При безрезультатности такого лечения - хирургическая операция.
Незаращение верхней губы (заячья губа)
Несращение боковых частей верхней губы с ее средней частью. Может быть односторонним и двусторонним. Затрудняет сосание
Хирургическая операция в первые месяцы жизни
Незаращение неба (волчья пасть)
Незаращение верхней челюсти и твердого неба, в результате чего получается расщелина, соединяющая полости рта и носа. Вызывает нарушение питания (попадание пищи в дыхательное горло, в полость носа), дыхания и речи. Часто сочетается с расщелиной в верхней губе
Хирургическая операция и протезирование; диспансерное наблюдение (смена лечебных аппаратов) до 16 лет
Полидактилия - многопалость, наличие лишних пальцев на кисти или стопе. Наиболее частый из врожденных пороков развития; чаще всего встречается в форме шестипалости, обычно на одной конечности.
Врожденный порок сердца
Неправильное внутриутробное формирование перегородки сердца (например, незаращение межпредсердной или межжелудочковой перегородки) либо сохранение после рождения особенностей внутриутробного кровообращения (например, открытый боталлов проток)
При незначительных дефектах межжелудочковой перегородки по мере роста сердца относительный размер отверстия уменьшается - вплоть до полного спонтанного закрытия. В других случаях - хирургическое лечение
Хотелось бы еще раз подчеркнуть, что когда речь идет о врожденных пороках развития, вопрос "Кто виноват?" не только непродуктивен, но и вреден, потому что отвлекает внимание от главного вопроса - "Что делать?". Поговорим на эту тему.
Что делать, если вы планируете беременность
- мужчины и женщины, в чьих семьях уже встречалось то или иное наследственное заболевание, - даже если сами они не больны
- семьи, где уже есть дети, страдающие врожденными пороками развития
- семьи, в которых предыдущие беременности заканчивались выкидышами или мертворождениями
- супруги, состоящие в родстве (например двоюродные и троюродные братья и сестры)
- женщины старше 35 и мужчины старше 50 лет
- мужчины и женщины, в связи со своим родом деятельности, состоянием здоровья или по каким-то иным причинам подвергающиеся воздействию перечисленных выше тератогенных факторов
Во всех этих случаях мы настоятельно рекомендуем партнерам, планирующим беременность, посетить медико-генетическую консультацию. Специалисты-генетики составят родословную, определят риск рождения ребенка с наследственным заболеванием. Нынешний уровень развития медицинских технологий позволяет сегодня в случае неблагоприятного прогноза прибегнуть к искусственному осеменению спермой донора или оплодотворению донорской яйцеклетки. Кроме того, следует по возможности исключить или свести к минимуму воздействие тератогенных факторов.
Что делать, если вы ждете ребенка?
Если вы ждете ребенка и входите в одну из перечисленных "групп риска". Первым шагом и в этом случае должен быть визит в медико-генетическую консультацию. Говорить об этом невесело, но бывают - хотя и очень редко - ситуации, когда на основании одной только родословной генетики приходят к заключению, что плод поражен заболеванием, несовместимым с жизнью. В таком случае, конечно, рекомендуется прерывание беременности. Однако, повторимся, случаи эти очень и очень редки. Как правило, специалисты медико-генетической консультации занимаются не диагностикой, а оценкой риска рождения ребенка с тяжелыми аномалиями и на основании этой оценки рекомендуют тот или иной метод пренатальной диагностики . Далее решение принимается в зависимости от результатов исследования. Насколько на самом деле высок риск родить ребенка с пороками развития, может решить лишь специалист. Не торопитесь делать аборт, если вы прочитали в аннотации, что лекарственный препарат, который вы принимали в самом начале беременности, не рекомендуется использовать в этот период; если принимали алкоголь, наркотики или перенесли острую респираторную вирусную инфекцию, сделали рентгеновский снимок на фоне беременности и т.п. Обязательно обратитесь в медико-генетическую консультацию, где сумеют правильно оценить реальный риск и порекомендуют необходимый комплекс исследований.
Что делать, если у вас родился ребенок с врожденным пороком развития
За окончательной медико-генетической консультацией по поводу прогноза на будущее лучше обратиться через 2-3 месяца, когда спадет психологическая напряженность и супруги смогут более объективно воспринимать такого рода информацию. Для большинства семей последующие беременности бывают успешными. Возможности пренатальной диагностики добавляют уверенности в благополучном исходе и врачам, и пациентам.
Имеются противопоказания. Ознакомьтесь с инструкцией или проконсультируйтесь у специалиста.

Пигментный ретинит именуемый по-другому абиотрофией сетчатки, представляет собой генетически обусловленное гетерогенное заболевание. Для него характерно изменение функции пигментного эпителия сетчатой оболочки глаза, что проявляется различными нарушениями зрения.
Клиническая картина пигментного ретинита и степень выраженности симптомов напрямую связаны с формой патологии. Особенно часто, проявлением заболевания становится ухудшение качества зрения со снижением его остроты и сужением полей. Кроме того, могут развиваться скотомы (слепые пятна) и нарушаться темновая адаптация. Во многих случаях пигментного ретинита, исходом заболевания становится слепота.
Для диагностики заболевания проводят сбор анамнеза и выполняют ряд офтальмологических исследований, включая осмотр глазного дна, электроретинографию, электроокулографию. Кроме того, назначают молекулярно-генетические анализы.
В клинической практике для пациентов с пигментным ретинитом применяют поддерживающую терапию, так как специфическое лечение заболевания (генная терапия с трансплантацией стволовых клеток), находится в стадии разработки.
Причины возникновения пигментного ретинита
Пигментный ретинит, известный также, как пигментная абиотрофия, является одним из дегенеративных заболеваний сетчатой оболочки глаза, наследственной природы. Данная патология сопровождается возникновением выраженных нарушений зрительной функции и чаще всего заканчивается полной слепотой.
На сегодняшний день выявлено несколько десятков генов, которые в сотнях вариантов мутаций, могут приводить к возникновению пигментного ретинита. Механизм наследования данного заболевания также различен. На данный момент описаны аутосомно-доминантные формы патологии, аутосомно-рецессивные, а также сцепленные с Х-хромосомой. К последним относятся, как рецессивные разновидности (носителями являются только мужчины), так и доминантные (когда заболевают представители обоих полов).
Встречаемость пигментного ретинита в популяции, в среднем составляет примерно 1 случай на 5 тысяч человек. Причем, некоторые формы заболевания встречаются чаще, другие – очень редко. Существуют данные медицинской статистики, согласно которым, почти 100-120 млн. человек на планете являются носителями генетических аномалий (включая бессимптомное носительство).
Вследствие генетической гетерогенности пигментного ретинита, этиология заболевание весьма разнообразна. Сегодня известно огромное количество его форм, что обусловлено мутациями различных генов. Однако, достоверно доказано, что причиной его возникновения становятся нарушения метаболизма в пигментном эпителии и фоторецепторах сетчатки, которые приводят к накоплению продуктов распада и вредных токсинов.
Классификация заболевания
Современная классификация пигментного ретинита включает четыре основные группы заболевания, подразделяющихся по механизму наследования генетического нарушения. Таким образом заболевание бывает:
- С аутосомно-доминантным путем наследования. Данный вариант патологии составляет 70-90% от всех случаев заболевания и считается наиболее распространенным. В качестве причин этой формы пигментной абиотрофии называют мутации генов PRPH2 (6 хромосома), IMPDH1 и RP9 (7 хромосома), RP1 (8 хромосома), а также широкого ряда других. Для всех известных генов характерна одна деталь — они кодируют белки, которые заняты в процессе метаболизма пигментного эпителия. Собственно, поэтому их структурные изменения приводят к различным расстройствам зрения. Пигментный ретинит с аутосомно-доминантным путем наследования, даже при большой его встречаемости, сопровождается менее выраженными нарушениями зрительных функций и медленным прогрессом. Поэтому, при адекватно выстроенной поддерживающей терапии заболевания, довольно часто удается значительно отсрочить наступление слепоты или даже избежать ее.
- С аутосомно-рецессивным путем наследования. Данная форма заболевания считается достаточно редкой и агрессивной. Проявления ее возникают довольно рано, в молодом или даже детском возрасте, а течение бывает столь быстрым, что полная слепота наступает еще до достижения человеком возраста взросления. Причиной аутосомно-рецессивного пигментного ретинита называют мутации в генах CRB1 (1 хромосома) и SPATA7 (14 хромосома). Вместе с тем, существуют более редкие формы заболевания, возникающие на фоне дефектов других генов. Аутосомно-рецессивные формы заболевания, пока остаются с недостаточно изученным патогенезом. Однако по имеющимся предположениям, кодируемые вышеуказанными генами белки, принимают участие в процессе эмбрионального развития органа зрения человека.
- С Х-сцепленным путем наследования.Эта генетическая форма заболевания признается специалистами одной из самых тяжелых. Как правило, она возникает при дефектах генов RP2 и RPGR имеющих рецессивный путь наследования. Этим обусловлено поражение такого типа формой заболевания исключительно мальчиков, у которых отсутствует гомологичная Х-хромосома. Названные выше гены задействованы в процессе кодирования белков-ферментов, активно участвующих в процессе метаболизма сетчатки глаза. В связи с этим, их дефект приводит к выраженным клиническим проявлениям заболевания.
- С мутациями митохондриальной ДНК. Данная форма пигментного ретинита очень редка. Заболевание наследуется исключительно по линии матери и передается потомству только от матери. Подвергающиеся мутации участки митохондриальной ДНК, врачам-генетикам при данной форме пигментного ретинита, пока выявить не удается.
Наряду с вышеописанными, существуют и иные типы классификаций данного заболевания: по клиническому течению, возрасту в котором возникли симптомы патологии (врожденный, ювенильный), наличию/отсутствию сопутствующих недостатков развития, и ряду прочих критериев. К настоящему моменту, общепринятая единая классификация пигментного ретинита отсутствует. Поэтому, разделение форм заболевания по механизму наследования, которому отдают предпочтение большинство офтальмологов, считается самым понятным и наиболее удобным. Только оно охватывает большинство генетических форм пигментного ретинита и их клинических особенностей.

Симптомы пигментного ретинита
Проявления пигментного ретинита могут возникать у носителя заболевания в любом возрасте. В раннем детстве, как правило, чаще развиваются симптомы рецессивных и сцепленных с Х-фактором форм патологи, а аутосомно-доминантные разновидности зачастую проявляют себя много позже – у людей зрелого и даже пожилого возраста.
Дальнейшее прогрессирование заболевания сопровождается ночной слепотой (никталопией), при том, что дневное зрение остается практически в норме. Это происходит от того, что в сетчатке дегенерационным изменениям подвергаются преимущественно фоторецепторы-палочки, задействованные в процессе световосприятия в условиях низкой освещенности.
Дистрофические изменения начинают постепенно затрагивать сосуды глаза, из-за чего фоторецепторы колбочки тоже разрушаются, хрусталик глаза и стекловидное тело мутнеют, склеры истончаются. Перечисленные процессы в совокупности и приводят к слепоте. Правда, такой исход присущ далеко не каждой форме заболевания. Для многих аутосомно-доминантных разновидностей пигментного ретинита характерно длительное время проявляющаяся дневная слепота (гемералопия) и сужение полей зрения невыраженного характера.
Диагностика заболевания
Если пациент отмечает снижение зрения в темное время суток, обязательно проведение тщательного офтальмологического осмотра. Так исследование глазного дна поможет обнаружить костные пятна – специфические точки по периферии сетчатки, которые являются ничем иным, как отложениями жироподобного пигмента. Прогрессирование заболевания приводит к увеличению их количества и продвижению в направлении макулы. На глазном дне, в случае выраженной картины заболевания, также выявляется атрофия капилляров и сужение артериол, а позднее — атрофия диска зрительного нерва восковидного характера.
При исследовании объема полей зрения, выявляется их концентрическое сужение, которое, в зависимости от стадии пигментного ретинита имеет различную степень выраженности.
Еще одним специфическим признаком данной патологии является снижение чувствительности (вплоть до тританопии) к синему цвету, что определяется с применением таблиц Рабкина.
Проведение электроокулографии ставит своей целью вычисление коэффициента Ардена, нормальное значение которого составляет 180% и более. При пигментном ретините, этот показатель находится на уровне 100% или еще более снижается.
Данные электроретинографии зависят от стадии пигментного ретинита. Таким образом, полученная картина может отображать состояние со снижением всех волн или даже нерегистрируемую ЭРГ, что присуще полной слепоте.
Молекулярно-генетические тесты – это последнее назначаемое исследование для окончательного постановления диагноза пигментного ретинита и определения его прогноза. Современным лабораториям доступны все методы генетической диагностики особенно распространенных форм заболевания, которые вызваны мутациями генов RP1, RP2, RPO, CRB1, SPATA7, RPGR и пр. Такие тесты информативны примерно для 70-80% случаев пигментного ретинита, однако при немногочисленных более редких формах заболевания они малоэффективны. В таких случаях, диагностическая техника, обычно сводится к секвенированию последовательности (прямому или автоматическому) вышеназванных генов.

Лечение пигментного ретинита и его прогноз
На сегодняшний день, разработка специфических методов лечения пигментного ретинита находится на стадии клинических испытаний и тестирования. В перспективе, для лечения заболевания планируется применять определенные методики генной терапии, с использованием стволовых клеток, а также другие эффективные медицинские методики.
На настоящий момент в офтальмологии практикуется только поддерживающее лечение, которое имеет своей целью замедление прогрессирования симптоматики заболевания. Для этого применяют витаминотерапию, препараты для улучшения трофики сетчатой оболочки и других глазных сред. В странах запада, больным с пигментным ретинитом могут предложить имплантацию протеза сетчатки, что весьма положительно отражается на зрительной функции. Правда, во многих случаях заболевания, особенно при его аутосомно-рецессивных и Х-сцепленных формах, вопреки всем терапевтическим мероприятиям, исходом пигментного ретинита становится полная слепота.
В связи с этим, прогноз пигментного ретинита, в целом считается неблагоприятным, ведь заболевание прогрессирует постепенно и неуклонно, в конечном итоге приводя к слепоте. При этом, различные формы пигментного ретинита отличает лишь скорость нарастания симптомов. У аутосомно-рецессивных вариантов она значительно выше, чем у доминантных форм патологии. Применение поддерживающей терапии, как правило, дает отсрочку в наступлении слепоты примерно на 5-10 лет, однако иных способов лечения пигментного ретинита в настоящее время нет.
В качестве профилактики пигментного ретинита может выступать медико-генетическая консультация родителей, относящихся к группам риска (уже имеющие симптомы заболевания или с наличием пигментного ретинита у близких родственников). По существующим данным регулярная защита глаз с помощью солнцезащитных очков, замедляет прогрессирование патологии, поэтому их использование также может быть предложено в качестве превентивной меры.
Она встречается почти у 80 миллионов людей в мире, а в России такой диагноз стоит у 1 500 000 людей, при этом такое же количество больных не подозревает о наличии у них заболевания. Открытоугольная форма глаукомы диагностируется в 70% случаев, поэтому о ее симптоматике, лечении и профилактике полезно знать каждому.
Патологические изменения при глаукоме
Радужка разделяет глаз на две камеры: переднюю и заднюю. Между камерами постоянно циркулирует жидкость, которая продуцируется цилиарным телом и выводится дренажным аппаратом. Сохраняющийся за счет постоянного передвижения баланс жидкости обеспечивает нормальное внутриглазное давление (ВГД). Если отток нарушается, давление начинает расти, и у пациента развивается глаукома.
Нарушение работы дренажной системы глаза происходит в результате:
- блокировки доступа к путям оттока;
- внутрисосудистых изменениях в путях оттока.
В первом случае речь идет о закрытоугольной глаукоме, которая встречается всего в 10-20% случаев. Если доступ к эвакуации жидкости был заблокирован корнем радужной оболочки, речь идет об ангулярной ретенции, а если причиной нарушения оттока выступает чужеродная ткань в фильтрующей зоне – о претрабекулярной ретенции.
Если пути оттока открыты, но дренажная функция глаза нарушена, у пациента диагностируется открытоугольная глаукома (ОУГ). При поражениях путей оттока внутренней стенки Шлеммова канала выявляют трабекулярную ретенцию, па при поражении наружной стенки – интрасклеральную ретенцию.
Внутриглазное давление любой этиологии сдавливает нерв, поэтому зрение неуклонно снижается. Без лечения, направленного на замедление прогрессирования болезни, человек полностью утрачивает зрение за 3-4 года.
Первичная ОУГ: причины и факторы риска
Глаукома – полиэтиологическое заболевание, которое может развиться под влиянием многих причин. Если среди нет травм или заболеваний органов зрения, которые могли привести к повышению давлению внутри глаза, глаукому считают первичной.
Существует три теории, способные объяснить патогенез открытоугольной формы глаукомы.
- Ретенционная теория – выделяет роль дистрофических изменений трабекулярного аппарата. Из-за затрудненного оттока влаги давление внутри глаза растет, и показатели офтальмотонуса выходят за пределы допустимых значений. Высокое ВГД вызывает ишемию диска зрительного нерва и гибель клеток сетчатки.
- Сосудистая теория – отмечает в качестве причины заболевания гемодинамические нарушения в сосудах глаза. Происходящие деструктивные изменения провоцируют гибель зрительного нерва и деформацию его многослойной решетчатой пластинки.
- Метаболическая теория – указывает на влияние пептидов, свободных радикалов и др. на развитие болезни.
Факторами, повышающими риск столкнуться с заболеванием и осложняющими его течение, являются:
- возраст старше 40 лет;
- отягощенная наследственность;
- другие заболевания органов зрения;
- близорукость и ранняя дальнозоркость;
- сильная пигментация трабекулярного аппарата.
Неблагоприятными признаками служат:
- значительные колебания ВГД в течение дня;
- разница показателей ВГД обоих глаз более 5 мм рт. ст.
Важно учитывать, что небольшие суточные изменения давления внутри глаза являются нормой: максимальные показатели фиксируют утром, после пробуждения, минимальные – вечером и ночью. Разница между давлением правого и левого в пределах 4 мм рт. ст.глаза тоже встречается у каждого человека.
Классификация ПОУГ
Открытоугольную глаукому можно классифицировать по разным признакам. Распространенным критерием является стадия развития патологии:
- начальная – повышение ВГД выше 21 мм рт. ст., постепенное сокращение полей зрения;
- развитая – истончение тканей зрительного нерва;
- далеко зашедшая – поражение зрительного нерва, значительное сужение полей зрения;
- терминальная – гибель зрительного нерва.
ОУГ можно классифицировать и по другим признакам:
- по уровню давления: нормальное, умеренно повышенное и высокое;
- по динамике развития: стабилизированная и нестабилизированная.
Но наиболее актуальным является классификация по механизму прогрессирования глаукомы, которая выделяет простую, эксфолиативную и пигментную форму, а также глаукому нормального давления.
Эксфолиативная открытоугольная глаукома
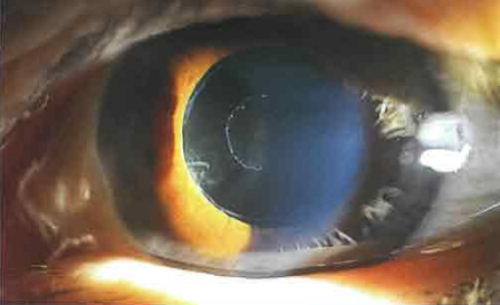
У пациентов с глаукомой на фоне псевдоэксфолиативного синдрома диагностируется эксфолиативная ОУГ. Данная форма характеризуется:
- двусторонним течением в 75% случаев;
- эксфолиацией хрусталика;
- дистрофией радужки;
- высоким ВГД.
Болезнь отличается неблагоприятным (быстрым) течением.
Пигментная открытоугольная глаукома
Для данной формы характерны следующие признаки:
- двустороннее течение;
- пигментация трабекулярной зоны;
- смещение периферической части радужки вперед;
- повышение давления внутри глаза более 26 мм рт.ст.;
- вымывание пигмента радужки или его перераспределение.
Активное высвобождение пигмента может происходить при выраженной физической нагрузке или при движении зрачка.
Глаукома нормального давления
При показателях ВГД, не выходящих за пределы нормы, врач может выявить:
- сужение полей зрения;
- поражение зрительного нерва;
- открытый угол передней камеры.
Сочетание признаков, указывающих на глаукому, с нормальным внутриглазным давлением, указывает на ГНД. Патологию можно рассматривать как подвид простой открытоугольной глаукомы с низкой чувствительностью зрительного нерва к высокому давлению внутри глаза.
Диагностика
Открытоугольная глаукома развивается незаметно: пациент не испытывает боли и другого дискомфорта до терминальной стадии болезни. Поэтому пациентам старше 40 лет нужно посещать офтальмолога превентивно каждые 2 года, чтобы вовремя выявить заболевание и принять меры, которые позволят сохранить зрение.
Поводом для внепланового обращения к врачу могут стать следующие симптомы:
- затуманивание зрения;
- изменение угла обзора;
- чувство тяжести в глазах;
- быстрая утомляемость органов зрения;
- радужные круги при взгляде на источник света.
Эти клинические признаки могут указывать на глаукому, а также на другие офтальмологические заболевания. Чтобы выяснить причину дискомфорта , подтвердить и уточнить диагноз, нужно выполнить диагностику.
1. Офтальмоскопия – осмотр глазного дна, который помогает оценить состояние диска зрительного нерва (ДЗН).
Во время исследования врач оценивает:
- размер ДЗН и неврального ободка;
- перипапиллярную атрофию;
- наличие кровоизлияний на ДЗН;
- состояние нервных волокон сетчатки.
При глаукоме у пациента происходят патологические изменения диска глазного нерва, которые можно зафиксировать во время исследования: экскавация, штрихообразные геморрагии по краю диска, побледнение зрительного нерва и др.

2. Гониоскопия – исследование угла передней камеры и радужной оболочки, позволяющее выяснить тип глаукомы (открыто- или закрытоугольная).
Во время процедуры врач использует местное обезболивание (капли), фиксирует голову пациента и осматривает его глаз, направив в него луч света. Осмотр позволяет увидеть характерные для ОУГ изменения, в том числе, пигментацию трабекулы.
3. Тонография – исследование внутриглазного давления, стабильное повышение которого является основным маркером прогрессирования глаукомы.
У здорового человека давление внутри глаз находится в пределах 11-21 мм рт. ст. На показатели может влиять возраст и время суток, на выраженное или постоянно фиксируемое в ходе диагностики повышение говорит о патологическом процессе.
Исследование также помогает оценить гидродинамику:
- минутный объем водянистой влаги (в норме от 1,5 до 4);
- коэффициент легкости оттока (в норме от 0,18 до 0,45);
- коэффициент Беккера – отношение ВГД к коэффициенту легкости оттока (норма менее 100).
В современной офтальмологии используют электронную тонографию.
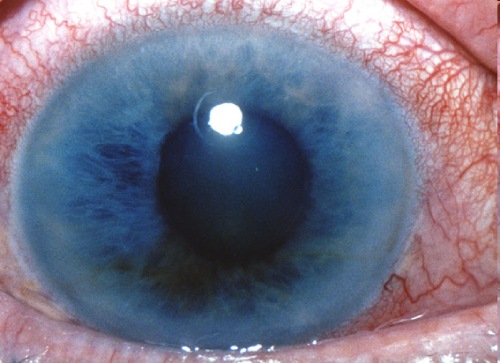
4. Биомикроскопия – безболезненный метод исследования, при котором врач использует специальный микроскоп и щелевую лампу.
При биомикроскопическом исследовании у пациентов с глаукомой выявляют:
- на конъюнктиве: сужение или расширение мелких сосудов, небольшие кровоизлияния, характерная извитость передних цилиарных артерий;
- на роговице: отек, отложение псевдоэксфолиаций на эндотелии;
- на радужке: пигментные отложения и депигментацию, неравномерность цвета глаз, атрофию;
- на хрусталике: помутнения и отложения псевдоэксфолиаций.
Глубина передней камеры глаза обычно остается без изменений.
Лечение
Глаукома требует пожизненного наблюдение у офтальмолога и непрерывную терапию для замедления прогрессирования болезни.
- После подтверждения диагноза врач подбирает препараты (капли), помогающие удерживать ВГД в пределах целевых показателей. Назначения корректируют 2-3 раза в год.
- Пациент регулярно проходит диагностику для оценки динамики состояния диска зрительного нерва.
По показаниям используются методики лазерных операций, эффективных при ОУГ:
- лазерная трабекулопластика;
- лазерная гониопластика;
- лазерная трабекулопунктура;
- лазерная десцеметогониопунктура;
- лазерная транссклеральная циклофотокоагуляция.
Также офтальмолог может назначить хирургическое вмешательство:
- фистулизирующего типа: трабекулэктомия, непроникающая склерэктомия, вискоканалостомия;
- дренажные: клапанные, аллодренажи, эксплантодренажи, дренажи из аутотканей;
- для сокращения секреции влаги: циклокриопексия, диатермокоагуляция задних длинных цилиарных артерий, лазерная циклофотокоагуляция.
Открытоугольная глаукома – серьезное заболевание, при котором человек должен постоянно наблюдаться у опытного офтальмолога и следовать всем его рекомендациям. В этом случае прогноз будет благоприятным и слепоты удастся избежать.

Х-сцепленное наследование принято делить на Х-сцепленное рецессивное и Х-сцепленное доминантное.
Х-сцепленное рецессивное наследование
Рис. 12. Родословная при Х-сцепленном рецессивном типе наследовании
Генетический риск. Если облигатная носительница Х-сцепленной рецессивной мутации вступает в брак со здоровым мужчиной, то каждый их сын будет иметь 50 %-й риск заболевания, а каждая дочь – 50 %-й риск быть носительницей. Поскольку мужчина передает хромосому Х только своим дочерям, а хромосому Y – сыновьям, то все дочери пораженных мужчин от браков со здоровыми женщинами являются облигатными носительницами, а все их сыновья здоровы. Таким образом, мужчина не может передать Х-сцепленное заболевание своему сыну за очень редким исключением при унипарентальной гетеродисомии.
В качестве примера Х-сцепленного рецессивного заболевания можно привести мышечную дистрофию Дюшена. Это самая частая мышечная дистрофия, первыми признаками которой являются переваливающаяся походка, трудности при подъеме по лестнице без болевых ощущений и тенденция к падениям ребёнка при ходьбе. Мышечная слабость
прогрессирует и пораженные мальчики умирают в конце второго – начале третьего десятилетия жизни. Таким образом, пораженные мужчины не имеют детей и не передают соответствующие мутации потомкам (рис. 13).
Рис. 13. Родословная семьи с мышечной дистрофией Дюшена
Вариабельная экспрессивность у женщин-гетерозигот. При многих Х-сцепленных болезнях женщины-гетерозиготы имеют мозаичный фенотип. Например, при Х-сцепленном альбинизме радужная оболочка и глазное дно больных мужчин не имеют пигмента, а у гетерозиготных женщин выявляется мозаичная (пятнистая) пигментация. Это объясняется феноменом Х-инактивации.
Х-сцепленное доминантное наследование
Х-сцепленные доминантные болезни являются редкими и выявляются у женщин-гетерозигот, а также у мужчин-гемизигот, имеющих мутантный аллель на единственной хромосоме Х. Х-сцепленное доминантное наследование напоминает аутосомно-доминантное. Но есть значимое отличие: пораженные мужчины передают заболевание только своим дочерям, а передача от отца к сыну невозможна (рис. 14). Примером этого типа наследования является витамин Д-резистентный рахит, при котором женщины обычно имеют более легкие формы заболевания, чем мужчины.
При многих Х-сцепленных доминантных болезнях у женщин может наблюдаться мозаицизм проявления болезни. Например, при синдроме Блоха – Сульцбергера (синдром недержания пигмента, тип II) наблюдается мозаичная пигментация кожи. Кроме того, это заболевание, также как синдром Ретта, является примером болезни, летальной для плодов мужского пола.
Рис. 14. Родословная при Х-сцепленном доминантном типе наследования
Наследование, сцепленное с хромосомой Y
Наследование, сцепленное с хромосомой Y, предполагает, что болеют только мальчики. Заболевание передается только от отца к сыну. В случае мутаций в генах хромосомы Y, вовлеченных в сперматогенез, возникает бесплодие вследствие азооспермии у мужчин. Технологии искусственного оплодотворения позволяют им иметь детей, но если при этом рождается сын, он также страдает азооспермией.
Влияние пола. Некоторые аутосомные признаки значительно чаще выявляются у одного из полов. Этот феномен получил название влияние пола. Лысина у мужчин является примером аутосомно-доминантного признака, ограниченного полом, что, по-видимому, является результатом влияния мужских половых гормонов. Другой пример – подагра, которая является очень редким состоянием у женщин до менопаузы, но после нее частота этого заболевания возрастает. При гемохроматозе
(аутосомно-рецессивном заболевании) у женщин-гомозигот намного реже возникает перегрузка железом и связанные с ней симптомы, чем у мужчин-гомозигот. Объяснением является физиологическая потеря железа женщинами во время менструаций.
Ограничение полом – это проявление определенных признаков у индивидуумов только одного пола. Пример: вирилизация девочек с аутосомно-рецессивным эндокринным заболеванием – врожденной гиперплазией коры надпочечников.
В табл. 4 кратко представлены основные признаки менделевских типов наследования.
Признаки менделевских типов наследования
Особенности заболевания у лиц разного пола
Особенности передачи
в родословной
Мужчины и женщины болеют в равной пропорции
Мужчины и женщины болеют в равной пропорции
Как правило, больны мужчины
Болеют мужчины и женщины с преобладанием женщин. Женщины поражены в меньшей степени, чем мужчины. В случае летальных для мальчиков болезней поражены только девочки, наблюдаются спонтанные аборты в семье.
Пораженные мужчины могут передавать заболевание своим дочерям, но не сыновьям. Передача от мужчины к мужчине исключает Х-сцепленный тип наследования.
Сцепленный с хромосомой Y
Болеют только мужчины.
Пораженные мужчины могут передавать заболевание только своим сыновьям.
Множественные аллели и комплексные признаки
Выше рассмотрены признаки, с которыми связаны только два аллеля – нормальный и мутантный. Некоторые гены имеют более двух аллельных форм, т.е. множественные аллели. Некоторые из них могут быть доминантными, другие – рецессивными по отношению к нормальному аллелю. Пример множественных аллелей – наследование групп крови человека.
Развитие генетики сделало возможным исследование комплексных признаков, которые формируются при взаимодействии нескольких генов. На этой основе возникла концепция олигогенного (дигенного и триаллельного) наследования.
При дигенном наследовании наблюдается аддитивный эффект гетерозиготных мутаций в двух различных локусах. Например, одна из форм пигментного ретинита, приводящая к потере зрения, вызвана гетерозиготностью по мутациям двух генов (ROM1 и periferin). Оба эти гена кодируют белки, присутствующие в фоторецепторах сетчатки глаза. Индивидуумы, гетерозиготные по мутации только одного из этих двух генов, не имеют клинических проявлений.
Триаллельное наследование можно рассмотреть на примере синдрома Барде-Бидля – редкого заболевания, характеризующегося ожирением, полидактилией, аномалиями почек, пигментным ретинитом и когнитивными нарушениями. Семь различных генных локусов, мутации в которых ведут к синдрому Барде–Бидля, были идентифицированы. До недавнего времени считалось, что заболевание наследуется аутосомно-рецессивно. Однако, сейчас известно, что есть одна форма синдрома, когда индивидуум, гомозиготный по мутациям одного локуса, является также гетерозиготным по мутации другого локуса. Таким образом, для того, чтобы заболевание проявлялось, необходимо три мутантных аллеля.
Антиципация. При некоторых аутосомно-доминантных болезнях манифестация симптомов более ранняя и течение болезни более тяжелое у потомков по сравнению с их родителями, также страдающими этим заболеванием. Феномен увеличения тяжести болезни из поколения в поколение называют антиципацией. Одним из объяснений антиципации является экспансия нестабильных триплетных повторов. В качестве примеров можно привести такие болезни экспансии триплетных повторов как миотоническая дистрофия, хорея Гентингтона, болезнь Кеннеди.
Читайте также:
- Опыт истории показывает что тоталитарные режимы заинтересованы в превращении граждан
- Оттаивание мерзлого грунта можно осуществлять методом
- Длинный венчик табака наследуется как рецессивный моногенный признак если в идеальной популяции
- Практикум что такое право
- Для чего предназначены правовые бд приведите примеры