
Как отозвать регистрационное удостоверение на медицинское изделие
Обновлено: 17.05.2024
19 марта 2020 года был введен особый порядок регистрации некоторых видов медицинских изделий, необходимых для предупреждения распространения инфекционных заболеваний. Упрощение процедуры регламентировано Постановлением Правительства № 299 от 18 марта 2020 г.
Правительство Российской Федерации упростило процедуру регистрации для 36 наименований медизделий – респираторов, медицинских масок, изолирующих халатов и костюмов, а также перчаток и бахил. Итоговый документ, регистрационное удостоверение, на данную продукцию ранее оформлялось в течении 3-5 месяцев. Теперь же, Росздравнадзор будет принимать решение о регистрации медицинских изделий, включенных в перечень, в течение трех-пяти рабочих дней дней после поступления первичного пакета документов. Далее уже потребуется в течении 150 дней довезти полный комплект документов.
Росздравнадзор опубликовал методические рекомендации по регистрации медицинских изделий с низкой степенью потенциального риска их применения.
Этим постановлением введен еще один вариант упрощенной регистрации (для продукции указанной в перечне приложенном к постановлению). В случае применения данного способа Регистрационное удостоверение действует до 1 января 2020 года, присутствует возможность регистрации партии медицинских изделий, признании иностранных протоколов испытаний.
В перечень к ПП 430 вошли аппараты искусственной вентиляции лёгких (аппарат ИВЛ) разных видов, оксигенаторы, перчатки, костюмы изолюрующие, халаты медицинские, маски разных видов, термометры и т.д. (рекомендуем ознакомиться с полным перечнем отраженным в постановлении).
Однако вместе с упрощением, введенным указанным выше постановление, введены ограничительные меры Постановлением Правительства № 433 от 03 апреля 2020 года по продаже изделий для предотвращения заражения и производных (маски, респираторы, перчатки медицинские, марля) на 90 дней.
Пояснения к ПП 430 и 433

Итого была упрощена процедура получения регистрационного удостоверения на:
1. Костюм изолирующий
2. Перчатки смотровые/процедурные из латекса гевеи,
неопудренные, нестерильные
3. Перчатки смотровые/процедурные из латекса гевеи,
опудренные
4. Халат операционный одноразового использования
5. Халат операционный многоразового использования
6. Халат изолирующий многоразового использования
7. Халат изолирующий одноразового использования
8. Маска лицевая для защиты дыхательных путей
многоразового использования
9. Перчатки смотровые/процедурные из полихлоропрена,
неопудренные
10. Перчатки смотровые/процедурные из полихлоропрена,
опудренные
11. Бахилы токонепроводящие, нестерильные
12. Халат для пациента одноразового использования
13. Халат для пациента многоразового использования
14. Халат процедурный одноразового использования
15. Халат процедурный многоразового использования
16. Респиратор общего применения
17. Костюм хирургический изолирующий
18. Маска хирургическая многоразового использования
22. Перчатки смотровые/процедурные нитриловые,
опудренные
23. Костюм хирургический на манжетах
24. Перчатки смотровые/процедурные виниловые,
неопудренные
25. Перчатки смотровые/процедурные виниловые,
опудренные
26. Костюм хирургический одноразового использования,
нестерильный
27. Бахилы водонепроницаемые
28. Бахилы токопроводящие, нестерильные
29. Перчатки смотровые/процедурные из гваюлового
латекса, неопудренные
30. Перчатки смотровые/процедурные из
этиленвинилацетата, неопудренные, нестерильные
31. Перчатки смотровые/процедурные нитриловые,
неопудренные, антибактериальные
32. Перчатки смотровые/процедурные полиизопреновые,
неопудренные
33. Набор одежды хирургический/смотровой
34. Перчатки смотровые/процедурные полиизопреновые,
опудренные
35. Перчатки смотровые/процедурные из латекса гевеи,
неопудренные, нестерильные, антибактериальные
36. Маска лицевая для защиты дыхательных путей
одноразового использования
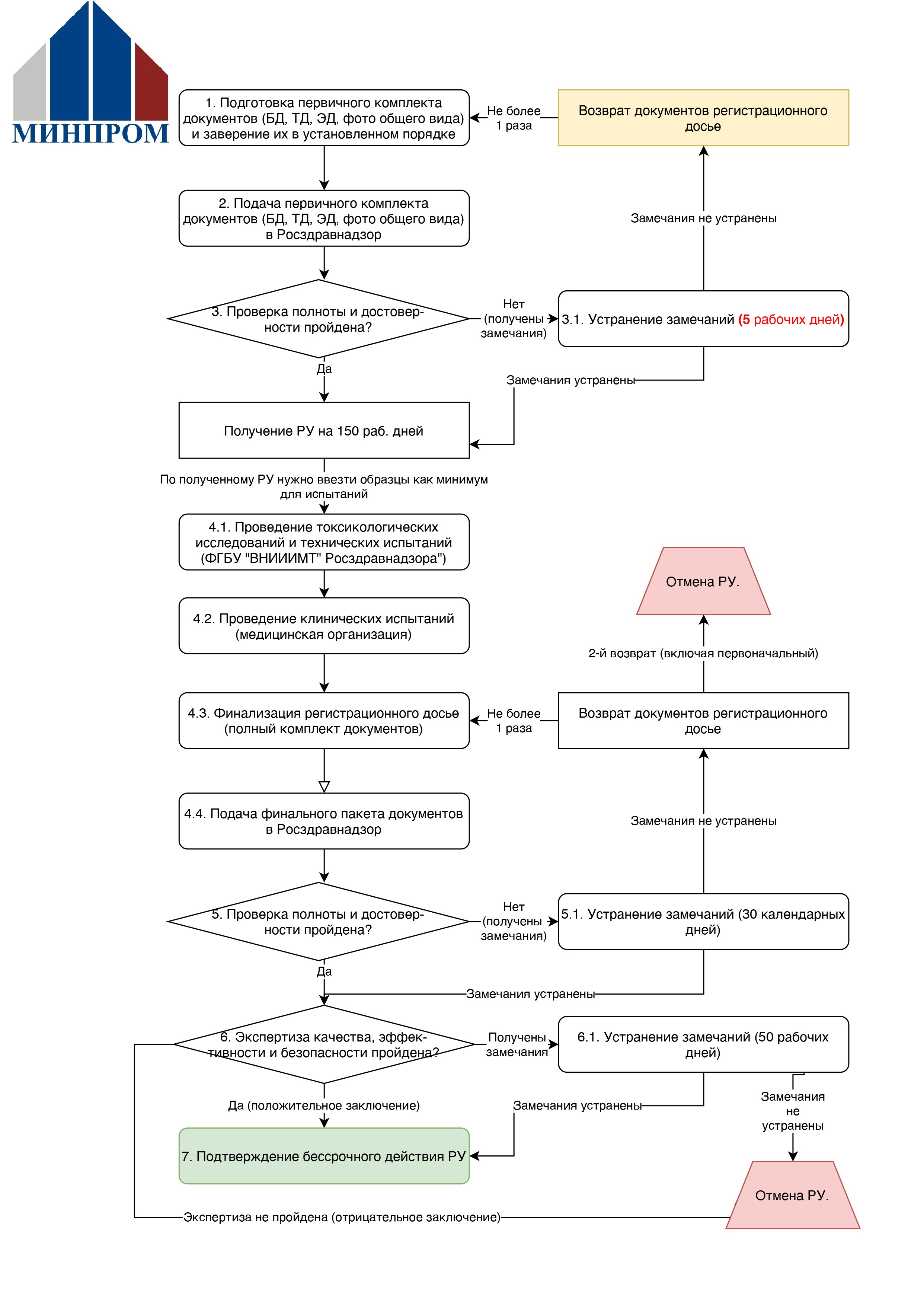
Процесс упрощенной регистрации
1) Подготавливается комплект базовых (юридических) документов, техническая и эксплуатационная документация, фото общего вида.
2) Данный пакет документов подается в Росздравнадзор.
3) Производится проверка полноты и достоверности, по результатам которой может выйти уведомление об устранении нарушений (необходимо устранить в течение 5 рабочих дней) или РУ.
4) В течение 150 рабочих дней во избежание аннулирования РУ необходимо провести токсикологические исследования, технические испытания, клинические испытания, доработать, при необходимости, техническую и эксплуатационную документацию, и подать пакет документов в Росздравнадзор.
5) Росздравнадзор проводит проверку полноты и достоверности, по результатам которой может выйти уведомление об устранении нарушений (необходимо устранить в течение 30 календарных дней) или направление в экспертное учреждение.
6) Экспертное учреждение проводит экспертизу в стандартном порядке (аналогично процедуре экспертизы при обычной регистрации медицинских изделий 1-го класса риска), по результатам которой выходит положительное заключение, замечания (необходимо устранить в течение 50 раб. дней) или отрицательное заключение (влечет отмену государственной регистрации и необходимость регистрации по стандартной процедуре.
7) После положительного заключения РУ продолжает действовать бессрочно.
Какие документы необходимы для оформления РУ
При регистрации медицинского изделия потребуется доверенность
Важно помнить что для регистрации иностранного медицинского изделия потребуется копия документа, подтверждающего полномочия уполномоченного представителя производителя (доверенность). Документ должен быть заверен либо апостилем либо в российском консульстве в стране изготовителя (с заверением полномочий подписанта). Рекомендуем ознакомиться с текстом и примером доверенности для оформления регистрационного удостоверения (подтверждение статуса уполномоченного представителя производителя)
Итого минимальный комплект документов требуемый для оформления составляет:
В дальнейшем потребуется довезти (в течении 150 дней):
Об особенностях обращения медицинских изделий, в том числе государственной регистрации серии (партии) медицинского изделия
ПРАВИТЕЛЬСТВО РОССИЙСКОЙ ФЕДЕРАЦИИ
ПОСТАНОВЛЕНИЕ
от 3 апреля 2020 г. № 430
МОСКВА
Об особенностях обращения медицинских изделий, в том числе государственной регистрации серии (партии) медицинского изделия
(В редакции постановлений Правительства Российской Федерации от 02.06.2020 № 804, от 13.11.2020 № 1826, от 06.03.2021 № 337, от 10.12.2021 № 2250)
В соответствии с частью 51 статьи 38 Федерального закона "Об основах охраны здоровья граждан в Российской Федерации" и пунктом 2 части 1 статьи 17 Федерального закона "О внесении изменений в отдельные законодательные акты Российской Федерации по вопросам предупреждения и ликвидации чрезвычайных ситуаций" Правительство Российской Федерации постановляет:
1. Утвердить прилагаемые особенности обращения медицинских изделий, в том числе государственной регистрации серии (партии) медицинского изделия.
2. Настоящее постановление вступает в силу со дня его официального опубликования и действует до 1 января 2025 г. (В редакции постановлений Правительства Российской Федерации от 13.11.2020 № 1826, от 10.12.2021 № 2250)
Председатель ПравительстваРоссийской Федерации М.Мишустин
УТВЕРЖДЕНЫ постановлением Правительства Российской Федерации от 3 апреля 2020 г. № 430
ОСОБЕННОСТИ ОБРАЩЕНИЯ медицинских изделий, в том числе государственной регистрации серии (партии) медицинского изделия
(В редакции постановлений Правительства Российской Федерации от 02.06.2020 № 804, от 13.11.2020 № 1826, от 06.03.2021 № 337, от 10.12.2021 № 2250)
1. Настоящий документ применяется при обращении медицинских изделий, которые предназначены для применения в условиях военных действий, чрезвычайных ситуаций, предупреждения чрезвычайных ситуаций, профилактики и лечения заболеваний, представляющих опасность для окружающих, заболеваний и поражений, полученных в результате воздействия неблагоприятных химических, биологических, радиационных факторов, по перечню согласно приложению № 1 (далее - медицинское изделие), в том числе государственной регистрации серии (партии) медицинского изделия. (В редакции Постановления Правительства Российской Федерации от 02.06.2020 № 804)
2. Регистрационное удостоверение на серию (партию) медицинского изделия выдается со сроком действия до 1 января 2025 г. (В редакции постановлений Правительства Российской Федерации от 13.11.2020 № 1826, от 10.12.2021 № 2250)
3. Для государственной регистрации медицинского изделия разработчик, производитель (изготовитель) медицинского изделия, уполномоченный представитель производителя (изготовителя) или лицо, осуществляющее ввоз медицинского изделия в Российскую Федерацию в целях его государственной регистрации (далее - заявитель), представляет либо направляет в федеральные государственные бюджетные учреждения, находящиеся в ведении Федеральной службы по надзору в сфере здравоохранения (далее - экспертное учреждение), в форме электронного документа и (или) на бумажном носителе:
заявление о государственной регистрации медицинского изделия в соответствии с требованиями пункта 9 Правил государственной регистрации медицинских изделий, утвержденных постановлением Правительства Российской Федерации от 27 декабря 2012 г. № 1416 "Об утверждении Правил государственной регистрации медицинских изделий" (далее - Правила регистрации);
копия документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя) (при наличии);
документы, подтверждающие принадлежность серии (партии) медицинского изделия заявителю на законных основаниях;
техническая документация производителя (изготовителя) на медицинское изделие (при наличии);
эксплуатационная документация производителя (изготовителя) на медицинское изделие, соответствующая требованиям, утвержденным Министерством здравоохранения Российской Федерации;
фотографические изображения общего вида медицинского изделия вместе с принадлежностями, необходимыми для применения медицинского изделия по назначению (размером не менее 18 на 24 сантиметра);
документы, подтверждающие результаты технических испытаний медицинского изделия, токсикологических исследований медицинского изделия, использование которого предполагает наличие контакта с организмом человека, клинических испытаний медицинского изделия, проведенных в соответствии с типовой программой испытаний в зависимости от вида медицинского изделия, разработанной экспертным учреждением (далее - типовая программа), и опубликованной на официальных сайтах экспертных учреждений в информационно-телекоммуникационной сети "Интернет" (по применимости). В случае проведения в Российской Федерации испытаний (исследований) по программе, отличной от типовой, экспертным учреждением определяется достаточность таких исследований (испытаний) для целей государственной регистрации серии (партии) медицинских изделий в соответствии с настоящим документом; (В редакции Постановления Правительства Российской Федерации от 02.06.2020 № 804)
иные документы, характеризующие качество, эффективность и безопасность медицинского изделия (при наличии);
В случае если указанные документы составлены на иностранном языке, они представляются с заверенным заявителем переводом на русский язык.
Требования, установленные Министерством здравоохранения Российской Федерации, о проведении оценки соответствия медицинских изделий в форме технических испытаний, токсикологических исследований, клинических испытаний в целях государственной регистрации медицинских изделий, не являются обязательными для испытаний (исследований) медицинских изделий, включенных в перечень. Данное положение применяется до ликвидации угрозы возникновения чрезвычайной ситуации или ликвидации чрезвычайной ситуации. Медицинские изделия, не прошедшие указанную оценку соответствия, подлежат повторной регистрации в соответствии с законодательством Российской Федерации.
4. Экспертное учреждение в течение 3 рабочих дней со дня поступления документов, указанных в пункте 3 настоящего документа, проводит оценку их полноты, достаточности и комплектности, в том числе достаточности объема проведенных испытаний (исследований) медицинских изделий для целей государственной регистрации серии (партии) медицинских изделий в соответствии с настоящим документом, оформляет заключение о возможности (невозможности) государственной регистрации серии (партии) медицинских изделий по форме, установленной Министерством здравоохранения Российской Федерации для целей экспертизы качества, эффективности и безопасности медицинского изделия, и направляет его в Федеральную службу по надзору в сфере здравоохранения.
К заключению экспертного учреждения прилагаются документы, представленные заявителем в соответствии с пунктом 3 настоящего документа.
В случае недостаточности для вынесения экспертным учреждением заключения материалов и сведений, содержащихся в представленных заявителем документах, предусмотренных пунктом 3 настоящего документа, экспертное учреждение в течение 2 рабочих дней со дня поступления указанных документов направляет заявителю заказным почтовым отправлением с уведомлением о вручении или в форме электронного документа, подписанного электронной подписью, либо в электронной форме по телекоммуникационным каналам связи запрос о представлении необходимых сведений с указанием характера замечаний и способа их устранения (далее - запрос). (Дополнен - Постановление Правительства Российской Федерации от 02.06.2020 № 804)
Заявитель обязан представить ответ на запрос экспертного учреждения в срок, не превышающий 5 рабочих дней со дня получения запроса. Экспертное учреждение готовит заключение в течение 2 рабочих дней со дня поступления от заявителя ответа на запрос. (Дополнен - Постановление Правительства Российской Федерации от 02.06.2020 № 804)
В случае непредставления по истечении 5 рабочих дней заявителем ответа на запрос экспертное учреждение подготавливает заключение на основании документов, имеющихся в его распоряжении. (Дополнен - Постановление Правительства Российской Федерации от 02.06.2020 № 804)
5. Заключение о невозможности государственной регистрации серии (партии) медицинских изделий оформляется экспертным учреждением при наличии следующих оснований (одного или нескольких):
а) отсутствуют доказательства соответствия медицинского изделия требованиям документации производителя (изготовителя);
б) отсутствуют доказательства безопасности медицинского изделия;
в) качество, и (или) эффективность, и (или) безопасность медицинского изделия не подтверждены полученными данными;
г) риск причинения вреда здоровью граждан и медицинских работников вследствие применения медицинского изделия превышает эффективность его применения;
д) медицинское изделие отсутствует в перечне, предусмотренном приложением № 1 к настоящему документу; (В редакции Постановления Правительства Российской Федерации от 02.06.2020 № 804)
е) документы, указанные в пункте 3 настоящего документа, не представлены в полном объеме.
6. Федеральная служба по надзору в сфере здравоохранения в течение 2 рабочих дней со дня поступления заключения, указанного в пункте 4 настоящих особенностей: (В редакции Постановления Правительства Российской Федерации от 02.06.2020 № 804)
б) принимает решение об отказе в государственной регистрации серии (партии) медицинского изделия и направляет заявителю мотивированный отказ заказным почтовым отправлением с уведомлением о вручении или в форме электронного документа, подписанного электронной подписью, либо в электронной форме по телекоммуникационным каналам связи (в случае поступления заключения о невозможности государственной регистрации медицинского изделия).
7. Регистрационное удостоверение на серию (партию) медицинского изделия оформляется в соответствии с требованиями пункта 56 Правил регистрации с указанием в нем дополнительно срока его действия, номера серии (партии) медицинского изделия, заводских номеров медицинских изделий (при наличии).
71. В отношении медицинских изделий, указанных в пунктах 1 - 18 приложения № 1 к настоящему документу, допускается внесение изменений в наименование медицинского изделия в части изменения сведений о его заводском номере (при наличии), номере серии (партии) в случае, если не изменились иные сведения, содержащиеся в документах регистрационного досье. (Дополнен - Постановление Правительства Российской Федерации от 02.06.2020 № 804)
72. Для внесения в документы, содержащиеся в регистрационном досье, изменений, указанных в пункте 71 настоящего документа, заявитель представляет (направляет) в Федеральную службу по надзору в сфере здравоохранения:
заявление о внесении изменений в документы, содержащиеся в регистрационном досье (далее - заявление о внесении изменений), оформленное в соответствии с пунктом 9 Правил регистрации;
копию документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя) (при наличии);
документы, подтверждающие принадлежность серии (партии) медицинского изделия заявителю на законных основаниях;
фотографические изображения общего вида медицинского изделия вместе с принадлежностями, необходимыми для применения медицинского изделия по назначению (размером не менее 18 на 24 сантиметра);
оригинал регистрационного удостоверения;
В случае если указанные документы составлены на иностранном языке, они представляются с заверенным заявителем переводом на русский язык.
(Дополнены пунктом - Постановление Правительства Российской Федерации от 02.06.2020 № 804)
73. Заявление о внесении изменений и документы, предусмотренные пунктом 72 настоящего документа, представляются заявителем в Федеральную службу по надзору в сфере здравоохранения на бумажном носителе непосредственно или направляются заказным почтовым отправлением с уведомлением о вручении и описью вложения или в форме электронного документа.
Федеральная служба по надзору в сфере здравоохранения принимает заявление о внесении изменений и документы, предусмотренные пунктом 72 настоящего документа, по описи.
(Дополнены пунктом - Постановление Правительства Российской Федерации от 02.06.2020 № 804)
74. В течение 3 рабочих дней со дня поступления заявления о внесении изменений и документов, предусмотренных пунктом 72 настоящего документа, Федеральная служба по надзору в сфере здравоохранения проводит проверку полноты и достоверности содержащихся в них сведений и осуществляет:
а) принятие решения о внесении изменений в документы, содержащиеся в регистрационном досье, которое оформляется приказом Федеральной службы по надзору в сфере здравоохранения, внесение в государственный реестр медицинских изделий и организаций (индивидуальных предпринимателей), осуществляющих производство и изготовление медицинских изделий, соответствующих сведений или принятие решения о возврате заявления о внесении изменений и документов, предусмотренных пунктом 72 настоящего документа, с мотивированным обоснованием причин возврата;
б) уведомление в письменной форме заявителя о принятом решении заказным почтовым отправлением с уведомлением о вручении с приложением переоформленного регистрационного удостоверения (в случае внесения изменений в него) и ранее выданного регистрационного удостоверения с отметкой о его недействительности (с указанием даты).
(Дополнены пунктом - Постановление Правительства Российской Федерации от 02.06.2020 № 804)
8. Федеральная служба по надзору в сфере здравоохранения формирует регистрационное досье на серию (партию) медицинского изделия с использованием следующих документов:
а) заявление о государственной регистрации и документы, предусмотренные пунктом 3 настоящего документа, а также заявление о внесении изменений и документы, предусмотренные пунктом 72 настоящего документа; (В редакции Постановления Правительства Российской Федерации от 02.06.2020 № 804)
б) заключение о возможности (невозможности) государственной регистрации серии (партии) медицинского изделия;
в) решение о государственной регистрации серии (партии) медицинского изделия, оформленное приказом Федеральной службы по надзору в сфере здравоохранения;
г) копия регистрационного удостоверения и копии уведомлений, оформленных Федеральной службой по надзору в сфере здравоохранения. (В редакции Постановления Правительства Российской Федерации от 02.06.2020 № 804)
9. Государственная пошлина за выдачу регистрационного удостоверения уплачивается в соответствии с законодательством Российской Федерации о налогах и сборах.
11. Федеральная служба по надзору в сфере здравоохранения принимает решение об отмене государственной регистрации серии (партии) медицинского изделия в соответствии с подпунктами "а" - "д" пункта 57 Правил регистрации.
____________
ПРИЛОЖЕНИЕ № 1к особенностям обращениямедицинских изделий, в том числе государственной регистрации серии (партии) медицинского изделия(в редакции постановленияПравительства Российской Федерацииот 10 декабря 2021 г. № 2250)
ПЕРЕЧЕНЬмедицинских изделий, которые предназначены для применения в условиях военных действий, чрезвычайных ситуаций, предупреждения чрезвычайных ситуаций, профилактики и лечения заболеваний, представляющих опасность для окружающих, заболеваний и поражений, полученных в результате воздействия неблагоприятных химических, биологических, радиационных факторов
Аппарат искусственной вентиляции легких стационарный высокочастотный с электроприводом
Аппарат искусственной вентиляции легких анестезиологический
Аппарат искусственной вентиляции легких высокочастотный с пневмоприводом портативный
Аппарат искусственной вентиляции легких высокочастотный с пневмоприводом для транспортировки пациентов
Аппарат искусственной вентиляции легких для интенсивной терапии неонатальный/для взрослых
Аппарат искусственной вентиляции легких для интенсивной терапии неонатальн
Судья Верховного Суда Российской Федерации Павлова Н.В., изучив жалобу общества с ограниченной ответственностью "Ангиолайн" (г. Новосибирск, далее общество, заявитель) на решение Арбитражного суда города Москвы от 10.03.2015 по делу № А40-177251/14, постановление Девятого арбитражного апелляционного суда от 26.06.2015 и постановление Арбитражного суда Московского округа от 13.10.2015 по тому же делу по заявлению общества о признании незаконными действий (бездействия Федеральной службы по надзору в сфере здравоохранения (далее Росздравнадзор), выразившихся в неприобщении изменений в ТУ, неуведомлении заявителя об отказе в приобщении изменений в ТУ, предоставлении неполного пакета документов экспертам ФГБУ ВНИИИМТ, неуведомлении заявителя о результатах экспертизы, отказе в отмене письма Росздравнадзора от 29.05.2014 № 02И-731/14 "О медицинском изделии, не соответствующем установленным требованиям"; признании незаконным и отмене письма Росздравнадзора от 29.05.2014 № 02И-731/14 "О медицинском изделии, не соответствующем установленным требованиям", как вынесенного на основе экспертного заключения, выданного по результатам исследования неполного пакета документов по вине Росздравнадзора,
решением Арбитражного суда города Москвы от 10.03.2015, оставленным без изменения постановлением Девятого арбитражного апелляционного суда от 26.06.2015 и постановлением Арбитражного суда Московского округа от 13.10.2015, в удовлетворении требований отказано.
В жалобе заявитель просит судебные акты отменить, ссылаясь на нарушение норм права.
Согласно положениям части 7 статьи 291.6 Арбитражного процессуального кодекса Российской Федерации (далее – Кодекс) кассационная жалоба подлежит передаче для рассмотрения в судебном заседании Судебной коллегии Верховного Суда Российской Федерации, если изложенные в ней доводы подтверждают наличие существенных нарушений норм материального права и (или) норм процессуального права, повлиявших на исход дела, и являются достаточным основанием для пересмотра оспариваемых судебных актов в кассационном порядке.
Оснований для пересмотра принятых по настоящему делу судебных актов в судебном заседании Судебной коллегии по экономическим спорам Верховного Суда Российской Федерации не установлено.
Общество обратилось в адрес Росздравнадзора с заявлением о внесении изменений в регистрационное удостоверение на медицинское изделие, в связи со сменой юридического адреса.
Росздравнадзором обществу выдано новое регистрационное удостоверение от 06.03.2013 № ФСР 2009/05402 на медицинское изделие.
В целях улучшения потребительских свойств и расширения размерного ряда обществом в феврале 2012 внесены изменения в ТУ 9444-002-83540797-2008 (извещение от 15.02.2012 № 2 об изменении ТУ 9444-002-83540797-2008 направлено в Росздравнадзор).
Экземпляр изменений в ТУ (повторно) направлен обществом в Росздравнадзор с сопроводительным письмом (вх. Росздравнадзора от 22.11.2013 № ММ-44352).
Росздравнадзор проинформировал общество о том, что в представленных им документах новая редакция технических условий вводит новые материалы стентов, устанавливает требования к стентам и устройствам (устройства отсутствуют в действующей редакции технических условий), и поскольку класс в зависимости от потенциального риска применения определяется в соответствии с приказом Минздрава России от 06.06.2012 № 4н "Об утверждении номенклатурной классификации медицинских изделий" (издан в соответствии с частью 2 статьи 38 Федерального закона от 21.11.2011 № 323-ФЗ "Об основах охраны здоровья граждан в Российской Федерации а не по ГОСТ Р 51609, указанному обществом в извещении от 15.02.2012 № 2, приобщение указанных документов согласно пункту 55 Правил государственной регистрации медицинских изделий, утвержденных Постановлением Правительства Российской Федерации от 27.12.2012 № 1416, не представляется возможным.
Кроме того, Росздравнадзор в письме от 10.02.2014 № 01-2217/14 сообщил обществу, что внесенные изменения не существенны и не влияют на характеристики эффективности, безопасности и функциональности изделия.
В связи с поступлением от Территориального органа Росздравнадзора по Тюменской области материалов внеплановой выездной проверки ГБУЗ ТО "Областная клиническая больница", г. Тюмень в декабре 2013 года Росздравнадзор поручил ФГБУ "ВНИИИМТ" Росздравнадзора провести экспертизу качества, эффективности и безопасности отобранных образцов медицинского изделия.
По результатам проведенной экспертизы образцов медицинского изделия отраженным в экспертном заключении ФГБУ "ВНИИИМТ" Росздравнадзора от 27.12.2013 № 14/ГЗ-13-0334-024 установлено, что качество и маркировка представленных образцов не соответствует требованиям ТУ 9444-002-83-83540797-2008.
Росздравнадзором издано информационное письмо от 29.05.2014 № 02И-731/14 "Об обращении на территории Российской Федерации медицинском изделии, не соответствующем установленным требованиям" "Стенты коронарные баллонорасширяемые "СИНУС" на системе доставки Рос РТС А 3,5 x 9 LOT 121011-01, Rx РТСА 4,0 x 9 LOT 121011-01, Rx РТСА 3,5 x 9 LOT 121011-01, Rx РТСА w 3,0 x 23 LOT 131107-02, Rx РТСА 2,5 x 19 LOT 131112-01, выпускаемые по ТУ 9444-002-83540797- 2008", производства ООО "Ангиолайн", 630090, Россия, Новосибирская область, г. Новосибирск, ул. Инженерная, д. 18, регистрационное удостоверение № ФСР 2009/05402 от 06.03.2013, срок действия не ограничен.
29.05.2014 Росздравнадзор письмом № 02-10493/14 направил в адрес общества копию экспертного заключения и уведомил его о подготовке информационного письма с копиями результатов экспертизы.
Полагая, что принятые решения и совершенные действия Росздравнадзора являются незаконными и влекут неблагоприятные правовые последствия в виде нарушения его прав и охраняемых законом интересов, общество обратилось в суд заявлением по настоящему делу.
Оценив в соответствии с требованиями главы 7 Кодекса представленные сторонами доказательства, в их совокупности и взаимосвязи, исходя из фактических обстоятельств дела, учитывая экспертное заключение ФГБУ "ВНИИИМТ" Росздравнадзора от 27.12.2013 № 14/ГЗ-13-0334-024, руководствуясь Федеральным законом от 21.11.2011 № 323-ФЗ "Об основах охраны здоровья граждан в Российской Федерации", Положением о Федеральной службе по надзору в сфере здравоохранения, утвержденным Постановлением Правительства Российской Федерации от 30.06.2004 № 323, положениями Административного регламента Федеральной службы по надзору в сфере здравоохранения и социального развития по исполнению государственной функции по регистрации изделий медицинского назначения утвержденного приказом Минздравсоцразвития Российской Федерации от 30.10.2006 № 735 (который действовал на дату внесения изменений в ТУ исходя из того, что при этом, внесение изменений в технические условия медицинского изделия в любом случае является изменением сведений о таком изделии, а следовательно подлежит правовому регулированию в соответствии с указанными нормативным правовым актом и осуществляется Росздравнадзором, установив факт несоответствия медицинского изделия требованиям ТУ 9444-002-83-83540797-2008 и отсутствия доказательств обратного, суды отказали в удовлетворении требований.
Суды пришли к выводу о том, что, обществом избран ненадлежащий способ защиты права, поскольку он не приводит к восстановлению нарушенного права заявителя, носит абстрактный и неопределенный характер.
Обстоятельства данного спора и представленные доказательства были предметом рассмотрения судов.
Довод заявителя о том, что заключения экспертизы не может быть обжаловано и не является надлежащим доказательством, отклоняется.
Правовое значение заключения экспертизы определено законом в качестве доказательства, которое не имеет заранее установленной силы, не носит обязательного характера и подлежит оценке судом наравне с другими представленными доказательствами.
В случае возникновения сомнений в обоснованности заключения эксперта лица, участвующие в деле, в силу части 2 статьи 87 Кодекса могут заявить ходатайство о проведении повторной экспертизы.
Однако, как следует из судебных актов, с таким ходатайством заявитель не обращался, в связи с чем, в силу части 2 статьи 9 Кодекса он несет бремя наступления неблагоприятных последствий несовершения им самим процессуальных действий.
Доводы заявителя по существу направлены на переоценку выводов суда о фактических обстоятельствах дела и имеющихся в деле доказательствах сводятся к изложению обстоятельств дела, которые были предметом исследования и оценки судов, что не свидетельствует о допущенных ими нарушениях норм материального и процессуального права и не являются основанием для передачи заявления с делом на рассмотрение Судебной коллегии Верховного Суда Российской Федерации.
обществу с ограниченной ответственностью "Ангиолайн" в передаче заявления для рассмотрения в судебном заседании Судебной коллегии по экономическим спорам Верховного Суда Российской Федерации отказать Судья Верховного Суда Российской Федерации Н.В.

Не только оборудование, инструменты или материалы, используемые в медицине, подлежат регистрации. Определенное программное обеспечение также является медицинским изделием и по закону его тоже необходимо зарегистрировать.
- профилактики;
- диагностики;
- лечения;
- медицинской реабилитации заболеваний;
- мониторинга состояния организма человека;
- проведения медицинских исследований;
- восстановления, замещения, изменения анатомической структуры или физиологических функций организма;
- предотвращения или прерывания беременности.
Причем перечень не является исчерпывающим . В информационном письме от 30.12.2015 Росздравнадзор установил список программного обеспечения, которое относится медицинским изделиям (постарался внести ясность).
К примеру, сюда вошло ПО, которое предназначено производителем для управления работой медицинского оборудования и мониторинга за его функционированием, получения от оборудования диагностических данных, их накопления и расчета в автоматическом режиме, мониторинга функций организма человека и передачи полученных данных, для 3D-моделирования и т.д.
Тем не менее на практике возникает большое количество споров — почему нужно воспринимать конкретное программное обеспечение как медицинское изделие. Например, административные системы для регистратуры, учета пациентов и т.п. — относить к такому ПО или нет?
14 февраля 2020 года Росздравнадзор внес изменения в критерии отнесения медицинского программного обеспечения к медицинским изделиям. Таким образом, более ранние рекомендации от 30 декабря 2015 года отменяются и вступают в силу новые. Теперь критерии стали более точными.
Если кратко — программное обеспечение, предназначенное производителем для оказания медицинской помощи и (или) интерпретации медицинской информации, однозначно относится к медицинскому изделию. Если таких признаков нет, то и регистрации ПО не подлежит.
До 31 декабря 2021 года изготовители медицинской продукции имеют право ставить ее на учет по национальным правилам. С 1 января 2022 года вступит в силу процедура ЕАЭС. Изготовители, которые не успели получить РУ по старым правилам до истечения этого срока, должны будут проходить регистрацию на основании Решения Совета ЕАЭК от 12 февраля 2016 г. № 46. Позаботьтесь о регистрации заранее!
По закону № 323-ФЗ к обращению на рынке в России допускаются только медизделия, прошедшие госрегистрацию в установленном режиме . Это позволяет допускать в оборот только качественные и безопасные изделия (в том числе ПО).
ПО, относящееся к медицинским изделиям, подлежит техническим испытаниям, экспертизе и последующей государственной регистрации. Документом, подтверждающим прохождение процедуры является регистрационное удостоверение (РУ). Кому оно понадобиться:
- производителям;
- организациям, осуществляющим реализацию ПО;
- учреждениям и организациям непосредственно применяющим программное обеспечение;
- импортерам (если медизделие покупается за рубежом).
Выдача РУ — это завершающая стадия. До того, как стать обладателем удостоверения необходимо подготовить техусловия, пройти анализ продукта и экспертизу. Возможно, понадобится провести доработку документации, повторную экспертизу и процесс затянется.
Кроме того, медицинские изделия делятся по классам риска — от класса 1 (требуют меньше всего времени для выполнения проверок, так как соответствует самому низкому уровню риска) и до класса 3 (проверка может проходить до 12 месяцев).
А теперь представьте, что вы постарались соблюсти все регламенты, но неверно заполнили заявление или получили не все необходимые для регистрации документы и вам отказали? Придется начинать все с начала.
Чтобы такого не произошло, предлагаем воспользоваться услугами ГК Аттек .
Мы работаем в регистрационном консалтинге уже 20 лет, из них 8 лет в медицинском. У нас в штате 3 кандидата медицинских наук и десяток экспертов. Мы консультируем и предоставляем первичную информацию бесплатно.
- работу с нулевого этапа — начиная с составления регистрационного досье;
- оформление технической документации (на этом шаге совершается наибольшее число ошибок, мы поможем их избежать);
- подбор учреждения для обследований (от правильности их выполнения зависит одобрение заявления на медицинскую регистрацию изделия).
Мы проведем всю работу от формирования досье до получения регистрационного удостоверения. Вся документация составляется в соответствии с действующим российским законодательством.
Вы также можете заказать у нас отдельные виды услуг, например, разработку ТУ или оформление декларации о соответствии для регистрации изделия медицинского назначения.
С нами вы получите РУ с первого раза, ведь Аттэк имеет большой опыт получения регистрационных удостоверений Росздравнадзора.
Читайте также: