
С какой целью реакцию получения стирола осуществляют при разбавлении газов реакции водяным паром
Обновлено: 28.06.2024
До 90 % мирового производства стирола осуществляется газофазным дегидрированием этилбензола при 580-650 °С [44]. В качестве катализаторов применяются Fe203, промотированный Сг03, КОН (NaOH) или V205. Процесс проводится в адиабатическом или изотермическом режиме. В первом случае энергоносителем служит перегретый до 800-900 °С водяной пар, который смешивают с парами этилбензола в мольном соотношении (10-14): 1. Введение перегретого пара снижает парциальное давление углеводородов и смещает равновесие, при этом повышается степень конверсии этилбензола, уменьшается закоксован - ность катализатора и увеличивается срок его службы, снижается перепад температур по высоте реактора. Смесь поступает на катализатор с температурой 640 °С и уходит из зоны реакции при 580 °С. Изотермический процесс проводят в трубчатом реакторе при 580-610 °С, тепло подводят путем непрямого теплообмена реакционной массы с теплоносителем - дымовыми газами, расплавами солей.
С целью полезного использования теплоты конденсации водяного пара предлагается компримировать часть потока контактного газа и направлять в качестве теплоносителя колонны выделения рециклового этилбензола, т. е. использовать по принципу "теплового насоса’ ' [49].
Термической деполимеризацией кубовых остатков ректификации стирола возможно получение дополнительного количества стирола. Процесс рекомендуется проводить с водяным паром в присутствии водородсодержащего газа, имеющего в своем составе 0.4-3 % (мае.) этилена и 1-4 % (мае.) метана [50].
Продолжаются исследования по поиску более эффективных катализаторов дегидрирования этилбензола. Так, японская фирма "Nissan Gridler Catalyst Со." разработала катализатор, в состав которого входят оксиды Fe, К, Se, Мо и Mg на
носителе, позволивший снизить температуру процесса до 580 °С при конверсии этилбензола 60-70 %. Срок службы нового катализатора на один год больше по сравнению со старым катализатором [51].
Активность и селективность Fe203 значительно повышаются при добавлении К2С03 [52]. При использовании катализатора состава, % (мае.): Fe203 - 23.46, алюминат Са - 10.43, Мо03 - 0.87, Се203 - 10.43, К2С03 - 39.1, К2Сг207 - 0.87, CaS04 - 2.61,
I СаС03 - 10.43, NaOH -1.8 при массовом соотношении водяной
Пар : этилбензол = (1.2-1.5): 1, температуре 580-590 С, атмосферном давлении и объемной скорости 1 ч'1 конверсия этилбензола 50 % и селективность по стиролу 97.2 % [53].
Обнаружен синергетический эффект для каталитической системы Ti02-Fe20g-Zr02 при дегидрировании этилбензола по сравнению с бинарными катализаторами Ti02-Fe203 и Zr02-Fe203 [54]. Активным и высокоселективным катализатором получения стирола является ортованадат магния на носителе MgO при концентрации V на поверхности свыше 10 % (мае.) [55].
Для повышения активности экструдированного железооксидного катализатора предложено получать его из cx-FeO(OH), который в свою очередь получается из скрапа металла дегидратацией Fe203 • хН20, имеющего размер частиц около 2 мкм и не содержащего связанного сульфата [56].
Фирма "Mitsubishi Petrochemical Co., Ltd." на основе лицензии "UOP" разработала установку дегидрирования этилбензола, на которой выход стирола увеличен за счет отвода выделяющегося водорода из зоны реакции [57].
На промышленной установке дегидрирования этилбензола до стирола в Китае процесс осуществляется в двух последовательных адиабатических реакторах: в первом реакторе при 615- 635 °С и 0.07 МПа, во втором - при 620-640 °С и 0.05 МПа. Дегидрирование проводится в присутствии воды в массовом соотношении к этилбензолу (1.3-1.8):1 и при объемной скорости подачи жидкого этилбензола 0.4-0.5 ч1. Для получения 1 т стирола требуется 1072 кг этилбензола, конверсия составляет 65.96 %, селективность образования стирола - 97.2 % [59].
Применение адиабатических реакторов с радиальным вводом сырья для производства стирола позволяет снизить гидравлическое сопротивление слоя катализатора в 2 раза по сравнению с
Реакторами с аксиальным вводом сырья и увеличить объемную скорость этилбензола с 0.5 до 1 ч“1 [60].
Установлено, что С02 в присутствии нанесенного на цеолит железооксидного катализатора РеМа^вМ-б играет роль окислителя, резко повышает активность и устойчивость катализатора к зауглероживанию [61].
Окислительное дегидрирование этилбензола предложено проводить при мольном соотношении С02: этилбензол = (50-70): 1 и температуре 500-700 °С. При использовании в качестве катализатора Ге/С, промотированного 20-30 % (мае.) 1лЖ)3, выход стирола 40-45 % при селективности его образования более 90 % [62].
Окислительное дегидрирование этилбензола позволяет снизить температуру процесса до 450-550 °С. Так, при проведении процесса при 530 °С на У-М^-катализаторе при объемной скорости 19910 ч-1 конверсия этилбензола 55.9 %, селективность образования стирола 92 % [63].
При проведении окислительного дегидрирования этилбензола при 420 °С и атмосферном давлении в присутствии ультра - дисперсного порошкообразного железо-молибденового катализатора на а-А1203 конверсия этилбензола составляет более 80 % при селективности образования стирола 60-65 % [64].
При окислительном дегидрировании этилбензола в кипящем слое катализатора состава, % (мае.): К20 - 12.5, Ьа203 - 10, В1203 - 25, ТЮ2 - 52.5 - конверсия этилбензола составляет около 98 %, выход стирола - 84 % [65].
Активными и селективными катализаторами получения стирола окислительным дегидрированием этилбензола являются Ugs(V04)2 и Mg2V04, причем в присутствии водяного пара селективность по стиролу несколько повышается [66].
При окислительном дегидрировании этилбензола на катализаторе №-У-8Ь / у-А1203 при 600 °С, мольном соотношении этил - бензол : 02: Н20 = 1 : 0.5 : 16, времени контакта 1 с достигается конверсия этилбензола 58.2 % при селективности образования стирола 89.3 % [67]. Катализатор стабилен, в отличие от аналогичных катализаторов, не содержащих V, которые быстро за- коксовываются.
Компания "ВАвЕ АС разработала процесс окислительного дегидрирования этилбензола в нестационарных условиях [68]. После проведения реакции катализатор продувается азотом, окисляется воздухом и снова продувается азотом. При этом достигается высокая конверсия этилбензола (свыше 97%) при 500 °С и селективность образования стирола более 95 %.
Запатентован способ совместного получения стирола и п-ме- тилстирола дегидрированием смеси этилбензола и /г-этилтолуола при 580-640 °С, разбавлении алкилбензолов перегретым водя ным паром в массовом соотношении 1 : (2.5-3) в присутствия Ее-Сг-К-катализатора [69]. Такой совмещенный процесс позво ляет повысить селективность по сумме мономеров И СНИЗИТ1 удельный расход водяного пара [70].
TOC o "1-5" h z В промышленности нашел применение процесс совместногс получения стирола и оксида пропилена, состоящий из следующих стадий [71]: с
С6н5сн2снз —^ сбн5сн(00н)сн3 ----- ► 'а
На предприятии АО "Нижнекамскнефтехим" для повышения селективности образования гидропероксида бензола использовали разбавление газа-окислителя азотом: при содержании кислорода 16 % (об.) расход этилбензола на 1 т гидропероксида был снижен с 0.96 до 0.86 т [72].
Эпоксидирование пропилена проводится в каскаде реакторов смешения или в комбинированной системе из реакторов смешения и вытеснения. Выход пропиленоксида составляет 83-90 %. Далее эпоксидат разделяют ректификацией, выделяя рециркулирующий пропилен, пропиленоксид, рециркулирующий этил - бензол и фракцию, содержащую метилфенилкарбинол.
Дегидратацию метилфенилкарбинола обычно проводят в паровой фазе с у-А1203 в качестве катализатора при 260-315 °С в двухступенчатом адиабатическом реакторе с промежуточным подогревом реакционной смеси. Эта стадия может осуществляться в присутствии водяного пара и водородсодержащего газа при массовом соотношении метилфенилкарбинол : вода : Н2= 1 : (0.3-0.6): : (0.0004-0.001) [73]. Для снижения термополимеризации стирола используются ингибиторы, например смесь гидрохинона и
N-диоксимхинона [74], нитрофенолы, нитрозамещенные сульфокислоты [75] или нитроксильные соединения [76].
Для ингибирования полимеризации продуктов дегидратации метилфенилкарбинола предложено использовать смесь 3,5-ди- трет-бутил-4-окси-Ы,1Ч-диметилбензиламина, алифатической карбоновой кислоты и хинона в массовом соотношении 1 : (0.1-2): : (0.0001-1). В качестве хинонов эффективны 3,3',5,5'-тетра-трет - бутилдифенохинон, 3,3', 5,5'-тетра-треяг-бутилстильбенхинон, л-диоксимхинон или их смеси. Рекомендуемое количество смеси ингибиторов 100-2000 ppm на продукты дегидратации метилфенилкарбинола [77].
Помимо прямой дегидратации метилфенилкарбинола с образованием стирола параллельно получается дифенилдиэтиловый эфир, который либо дегидратируется до стирола, либо разлагается, давая ацетофенон и этилбензол [71]:
С6Н5СНОН --------- ► (С6Н5СН—)20 ---------- С6Н5—СН=СН2
TOC o "1-5" h z - н2 L >
------ ► С0Н5 С—О С6Н5СН2СН3
_____________ +н2, - н2о______________ J
Образующийся ацетофенон селективно гидрируют в присутствии медьсодержащих катализаторов, например суспендированного Си-Сг-Ва-катализатора, и стабилизирующей добавки для снижения удельного расхода катализатора - гексеналя, добавляемого в количестве 0.01-0.5 % (мае.) от ацетофенона [78]. Гидрированию подвергают обычно высококипящую фракцию, остающуюся после выделения стирола ректификацией. Конверсия ацетофенона составляет не менее 70%, а селективность - 95 % [79].
Остающиеся высококипящие продукты отделяют ректификацией и сжигают. Однако их можно утилизировать в присутствии в качестве катализатора А1203, модифицированного органическими и неорганическими соединениями [80, 81]. При 150- 400 °С конверсия высококипящих продуктов 50-96 %, выход стирола, ацетофенона, этилбензол а и толуола составляет в пересчете на метилфенилкарбинол 60-85, 10-25, 5-10 и 2-5 % соответственно.
Фирма "The Dow Chemical Co." разработала и испытала ш полупромышленной установке двухстадийный метод получение стирола из фракции С4 продуктов пиролиза, содержащей бутадиен бутены и бутан [82]. Фракцию С4 очищают от примесей ацетиленовых углеводородов селективным гидрированием, затем димери- зуют в 4-винилциклогексен при 100 °С и избыточном давлении 1.75 МПа на цеолитном катализаторе, содержащем соли одновалентной меди. На заключительной стадии 4-винилциклогексен смешивают с водяным паром и кислородом и в реакторе с оксидным катализатором при 400 °С и избыточном давлении 0.525 МПа получают стирол. Примеси кослородсодержащих соединений удаляют адсорбцией на А1203. Отмечается, что этот метод экономичнее дегидрирования этилбензола на 0.25-0.4 долл/кг стирола.
Стирол можно получать также метатезисом этилена со стиль- беном, полученным окислением толуола [83]:
С6Н5СНз + 02 —— - С6Н5СН=СНС6Н5+2Н20
С6н5сн=снс6н5 + сн2—сн2 ■ —ос ► 2С6Н5СН=СН2
Разработан процесс производства стирола дегидратацией
2- фенилэтанола, который может быть получен из толуола или из бензилового спирта [84]:
Та же фирма для газофазного дегидрирования 4-винилциклогексен а запатентовала катализатор, содержащий Sn и Sb на носителе - алюмосиликате, гипсе, MgO, ТЮ2 [85]. Катализатор снижает свою активность в 2 раза через 1000-2500 ч работы. При соотношении Sn : Sb = 9 : 1, температуре 380 °С, мольном соотношении вода : 4-винилциклогексен : 02: N2 = 14.4 : 1 : 1.506 : : 19.9 через 24 ч конверсия составляет 98 %, выход стирола 91 % при селективности его образования 93 %. Промотирование катализатора 1.5 % (мае.) Fe приводит к повышению селективности до 97.2 % и выхода стирола до 94.1 %.
1) Термическое декарбоксилирование коричной кислоты проводится при температуре 120-130 О С и атмосферном давлении. Выход стирола составляет около 40%

2) Дегидратация фенилэтилового спирта. Реакция может быть реализована как в газовой, так и в жидкой фазе. Жидкофазная дегидратация фенилэтилового спирта осуществляется в присутствии фосфорной кислоты или бисульфита калия. Дегидратация в паровой фазе проводится над катализаторами: оксидами алюминия, тория или вольфрама. При использовании оксида алюминия выход стирола составляет до 90% от теории.

3) Синтез из ацетофенона. Стирол можно получить по реакции ацетофенона с этиловым спиртом над силикагелем:

Выход составляет около 30%.
4) Получение стирола из галогенэтилбензола:


5) Получение стирола дегидрированием этилбензола.
6) Метод производства из этилбензола через гидропероксид этилбензола с одновременным получением оксида пропилена (халкон-процесс):
 | ||
 | ||
7)Получение стирола метатезисом этилена со стильбеном, полученным окислением толуола:
 |
8) Получение стирола каталитической циклодимеризацией бутадиена:

Все приведенные методы получения стирола (за исключением дегидрирования) многостадийны, используют повышенное давление и высокую температуру, что приводит к усложнению и удорожанию производства. Для некоторых методов используется не очень доступное сырье. Небольшие выходы.
Основным методом промышленного производства стирола является каталитическое дегидрирование этилбензола. Этим методом получают более 90% мирового производства этилбензола. В качестве катализаторов дегидрирования применяются сложные композиции на основе оксидов цинка или железа. Раньше наиболее распространенным был катализатор стирол-контакт на основе ZnO. В последнее время используют, главным образом, железо-оксидные катализаторы, содержащие 55-80% Fe2O3; 2-28% Cr2O3; 15-35% K2CO3 и некоторые оксидные добавки. В частности широко используется катализатор НИИМСК К-24 состава Fe2O3 – 66-70%; K2CO3 – 19-20%; Cr2O3 – 7-8%; ZnO2 – 2,4-3,0%; K2SiO3 – 2,0-2,6%. Значительное содержание K2CO3 в катализаторе обусловлено тем, что он способствует дополнительной саморегенерации катализатора за счет конверсии углеродистых отложений водяным паром. Катализатор работает непрерывно 2 месяца, после чего его регенерируют, выжигая кокс воздухом. Общий срок службы катализатора – 2 года.
Реакционный узел для дегидрирования этилбензола можно выполнять различными способами. Один из вариантов – трубчатый реактор, обогреваемый топочным газом по типу, изображенному на рисунке.

Его достоинство – близкий к изотермическому профиль температуры, что позволяет получать повышенную степень конверсии при хорошей селективности. Однако высокие металлоемкость и капитальные затраты на такой реактор привели к созданию других аппаратов – со сплошным слоем катализатора, не имеющих поверхностей теплообмена (рис.А).

Они работают в адиабатических условиях, и реакционная смесь постепенно охлаждается, причем водяной пар играет здесь и роль аккумулятора тепла, не давая смеси чрезмерно охладиться. При получении стирола в единичном адиабатическом реакторе обычная степень конверсии этилбензола составляет около 40%. Недостатки такого единичного реактора – существенное охлаждение смеси, одновременное смещение равновесия в нежелательную сторону и зависящее от этого снижение скорости и селективности. Степень конверсии нельзя довести до приемлемой величины, т.к. это повышает удельный расход пара. Другие установки (рис.Б) приближают процесс к изотермическому и лучше учитывают особенности равновесия реакции. В такой установке имеются 2 реактора (или два слоя катализатора). Охладившуюся в первом реакторе смесь до подачи во второй реактор нагревают перегретым паром. Реактор на рисунке В имеет два-три кольцевых слоя катализатора, причем в первый слой поступает весь этилбензол, но лишь часть водяного пара. В пространство между слоями катализатора подают дополнительное количество перегретого пара. С его помощью повышается температура смеси и происходит ступенчатое разбавление смеси с удалением ее от равновесного состояния, что способствует росту скорости и селективности реакции.

Совмещенное дегидрирование и окисление метанола. Получаемые и побочные продукты. Условия проведения процесса. Особенности оформления реакционного узла.
Дегидрированием или окислением первичных спиртов получают только формальдегид (из метанола). Формальдегид НСНО представляет собой в безводном состоянии бесцветный газ с острым раздражающим запахом (т. конд. -19ОС при 0,1 МПа). При хранении он легко полимеризуется и нередко выпускается в виде твердого полимера — параформальдегида (параформ), который легко деполимеризуется.
Большей частью формальдегид выпускают в виде 37%-ного водного раствора, называемого формалином. В нем формальдегид присутствует в виде гидрата НСНО•Н20 и низкомолекулярньтх полимеров (полиоксиметиленгликоли). Во избежание более глубокой полимеризации и выпадения осадка добавляют к формалину 7—12% (масс.) метилового спирта в качестве стабилизатора.
Совмещенное дегидрирование и окисление метанола. Дегидрирование первичных спиртов, в том числе метанола, менее благоприятно по сравнению с вторичными спиртами по условиям равновесия и селективности реакции. По этой причине, а также с целью устранения эндотермичности процесса осуществили совмещенное дегидрирование и окисление метанола:


Можно так подобрать соотношение этих реакций, чтобы суммарный тепловой эффект был только немного положительным, но достаточным для возмещения потерь тепла в окружающую среду и для нагревания исходной смеси до нужной температуры. Практически при получении формальдегида такое положение достигается, когда процесс на 55% идет через окисление и на 45% через дегидрирование, и тогда процесс можно осуществить в адиабатических реакторах, не имеющих поверхностей теплообмена. В этом состоит одно из преимуществ совмещенного процесса окисления и дегидрирования спиртов. При указанном соотношении реакций дегидрирования и окисления исходная паро-воздушная смесь должна содержать 45% (об.) метанола, что находится за верхним пределом взрываемости метанола в воздухе [34,7% (об.)].
При получении формальдегида кроме основных реакций протекают побочные процессы более глубокого окисления, дегидрирования и гидрирования, ведущие к образованию оксидов углерода, муравьиной кислоты, воды и метана:
Окислительное дегидрирование проводят при недостатке кислорода, поэтому глубокое окисление не получает значительного развития. В то же время само дегидрирование, инициируемое кислородом, протекает быстрее, и все ранее упомянутые побочные реакции не так заметны, как при дегидрировании первичных спиртов. Это позволяет работать при более высокой температуре (500—б00°С), большой скорости реакции и времени контакта 0,01—0,03 с. Выход формальдегида на пропущенное сырье достигает 80-85% при степени конверсии метанола 85-90%. Замечено, что добавление воды к исходному метанолу повышает выход и степень конверсии, по-видимому, в результате разложения ацеталей. Катализаторами синтеза формальдегида этим методом служит металлическая медь (в виде сетки или стружек) или серебро, осажденное на пемзе. Последний катализатор оказался более эффективным и широко применяется в промышленности.
Технологическая схема производства формальдегида окислительным дегидрированием метанола изображена на рис. 139. Метанол, содержащий 10-12% воды, из напорного бака 1 непрерывно поступает в испаритель 2. Туда же через распределительное устройство подают воздух, очищенный от пыли и других загрязнений. Воздух барботирует через слой водного метанола в нижней части испарителя и насыщается его парами. В 1 л образующейся паро-воздушной смеси должно содержаться 0,5 г метанола. Поддержание такого состава смеси очень важно для обеспечения взрывобезопасности и нормального протекания процесса. Поэтому работа испарительной системы полностью автоматизирована: поддерживают постоянные уровень жидкости в испарителе, ее температуру (48—50°С) и скорость подачи воздуха, благодаря чему обеспечиваются необходимые температурный режим и степень конверсии в адиабатическом реакторе.
Паро-воздушная смесь проходит брызгоуловитель, находящийся в верхней части испарителя, затем перегреватель З и поступает в реактор 4, в средней части которого находится катализатор. Реакционные газы сразу же попадают в подконтактный холодильпик 5 (смонтирован вместе с реактором), где происходит быстрое охлаждение смеси и предотвращается распад формальдегида. В разных схемах охлаждение осуществляют проточной водой или паровым конденсатом, когда холодильник играет роль генератора пара низкого, среднего или даже высокого давления. Полученный пар (или горячая вода) служит для перегрева постулающей смеси в теплообменнике 3 и для обогрева испарителя 2.
Охлажденные реакционные газы поступают в абсорбер 6, выполненный в виде тарельчатой колонны; жидкость на тарелках охлаждают внутренними или выносными холодильниками (на схеме не изображены). Абсорбер орошают таким количеством воды, чтобы в кубе получился 36—37%-ный формалина. Стадии абсорбции и разделения продуктов оформляют двумя разными способами.
По одному из них в абсорбере поглощают как форальдегид, так и непревращенный метанол, который содержится в продуктах реакции в количестве, как раз достаточном для стабилизации формальдегида. В этом случае верхнюю тарелку абсорбера охлаждают рассолом, а колонна 7 служит лишь для санитарной очистки газа, в то время как для получения безметанольного формалина (требуемого иногда для ряда целей) необходима установка для отгонки метанола. При втором способе в абсорбере поглощают преимущественно формальдегид; тогда колонна 7 служит для абсорбции метанола, который отгоняют от воды и возвращают на реакцию. В обоих случаях формалин из куба абсорбера б охлаждают в холодильнике 8 и собирают в сборнике 9.

Значительная эндотермичность дегидрирования обусловливает применение трубчатых реакторов, в межтрубном пространств которых циркулируют горячие газы от сжигания газообразного или жидкого топлива. Схема типичного реакционного узла дл дегидрирования спиртов представлена ниже. В топке З происходит сгорание топливного газа, подаваемого вместе с воздухом через специальные форсунки. Температура топочных газов слишком высока, поэтому их разбавляют обратным газом (циркуляция его в системе осуществляется газодувкой 4). Спирт поступает вначале в систему испарителей-перегревателей 1, где он нагревается до нужной температуры частично охлажденными топочными газами. Затем пары спирта попадают в реактор 2, где в трубе находится катализатор. Реакционная смесь подогревается горючими топочными газами, находящимися в межтрубном пространстве, что компенсирует поглощение тепла из-за эндотермичности процесса. По выходе из контактного аппарата реакционные га охлаждают в холодильнике-конденсаторе (на рисунке не показан), а в случае летучих продуктов их дополнительно улавливают водой. Полученный конденсат (и водные растворы) ректифицируют, выделяя целевой продукт и непрореагировавший спирт, возвращаемый на дегидрирование.

Однако, несмотря на многообразие известных способов, основным методом получения стирола в промышленных масштабах остается каталитическое дегидрирование этилбензола при высоких температурах.
Разработан способ получения стирола каталитическим дегидрированием этилбензола в присутствии водяного пара с последующей ректификацией углеводородного конденсата, который предварительно перед стадией ректификации подвергают каталитическому гидрированию на палладийсодержащих катализаторах при температуре 20-30 о С, объемной скорости 4,5-5,0 ч -1 и объемном соотношении водород-сырье, равном 35-45, для выделения стирола-ректификата, возвратного этилбензола, бензолтолуольной фракции и кубового остатка ректификации стирола углеводородный конденсат [22].
Наибольшую сложность в технологическом процессе выделения стирола представляет стадия его очистки от этилбензола и побочных продуктов, образующихся при дегидрировании [23, 24].
Технология совместного получения стирола и оксида пропилена состоит из следующих стадий:
- получение гидропероксида этилбензола (ГПЭБ) окисление этилбензола
- выделение гидропероксида этилбензола;
- эпоксидирование полученным гидропероксидом олефина (пропилена) и разделение продуктов эпоксидирования
- дегидратация образовавшегося метилфенилкарбинола (МФК) и выделение стирола
- гидрирование ацетофенона с образованием метилфенилкарбинола
Таким образом, это производство является многостадийным. Оно наиболее перспективно, потому что наряду со стиролом позволяет получать важные эпоксидные соединения, например, оксид пропилена. Стирол и оксид пропилена имеют большое значение в отрасли основного органического и нефтехимического синтеза. Если первый является важнейшим мономером для производства широкого круга каучуков и полимеров, то второй используется не только для производства каучуков (пропиленоксидных каучуков), но и важных продуктов и полупродуктов основного органического и нефтехимического синтеза (пропиленгликолей, неионогенных поверхностно-активных веществ - проксанолов и проксаминов, аллилового спирта, пропиленкарбоната, изопропаноламинов).
Конечно же, целесообразно совместное получение нескольких целевых продуктов, чем их получение с помощью самостоятельных процессов. Кроме того, такой способ получения продуктов не только экономически, но и экологически более чистый, так как в нем образуется меньше побочных продуктов, на выделение которых тратится энергия и сырье.
Поскольку дегидратация метилфенилкарбинола происходит при относительно невысоких температурах (~300 о С), то стирол получается с минимальным количеством примесей. Ещё одним преимуществом этого метода является более простая схема ректификации стирола.
В процессах получения стирола различными промышленными способами (дегидрированием этилбензола или через стадию дегидратации метилфенилкарбинола), образование побочных продуктов термодинамически вероятно на всех стадиях процесса. Однако наибольшую скорость образования побочных продуктов следует ожидать в реакторе дегидрирования, промежуточном межреакторном теплообменнике, на первой ступени конденсации контактного газа и в кипятильниках ректификационных колонн.
При высоких температурах в реакторе возможно протекание следующих реакций:
- дегидрирование с образованием стирола, фенилацетилена, дивинилбензола и водорода
При дегидрировании дибензила возможно образование стильбена
Путем конденсации образуются антрацен, фенантрен, нафталин, стильбен, алкилинданы и другие соединения [26].
При конденсации в продуктах процесса дегидрирования этилбензола в стирол возможно попадание воздуха, что может привести к образованию продуктов окисления стирола и этилбензола, а в процессе совместного получения стирола и оксида пропилена идет образование кислородсодержащих соединений: ацетофенона, бензойной кислоты, бензальдегида и др.
В основе технологии получения гидропероксида этилбензола лежит реакция жидкофазного окисления этилбензола молекулярным кислородом. В результате окисления этилбензола наряду с гидропероксидом (на него расходуется более 98 % этилбензола) образуются метилфенилкарбинол и ацетофенон. Кроме того, в продуктах окисления обнаружены бензальдегид, бензойная и муравьиная кислоты, пероксид водорода, 2,3-дифенилбутан.
В настоящее время установлено, что реакция получения гидропероксида этилбензола протекает по схеме, учитывающей параллельно-последовательный механизм образования ацетофенона и возможность рекомбинации радикалов RO2 непосредственно в МФК и ацетофенон (а не через стадию образования гидропероксида). Следовательно, схему окисления этилбензола можно представить следующим образом:
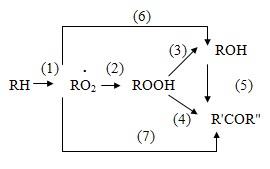
Результаты кинетических исследований реакции окисления этилбензола показывают, что скорости образования метилфенилкарбинола и ацетофенона по реакциям (3), (6) и (4), (7) равны между собой. Это свидетельствует о том, что скорости образования кетона и спирта через пероксидные радикалы одинаковы. Однако при накоплении в системе определенного количества метилфенилкарбинола скорость реакции (5) превышает суммарную скорость реакций (4) и (7), а метилфенилкарбинол становится основным источником образования ацетофенона [25, 27].
Кроме того, в процессе совместного получения стирола и оксида пропилена, возможно, протекание следующих последовательно-параллельных реакций:
- стадия эпоксидирования, на которой ГПЭБ и пропилен, реагируют с образованием оксида пропилена, образовавшийся при этом МФК является сопутствующим продуктом
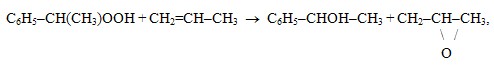
- реакция ГПЭБ и пропилена с образованием МФК и ацетона
- ГПЭБ и пропилен реагируют с образованием изопропилового спирта и ацетофенона
- ГПЭБ разлагается с образованием фенола и ацетальдегида
- ГПЭБ разлагается с образованием ацетофенона и воды
- дегидратация метилфенилкарбинола в стирол
- метилфенилкарбинол дегидрируется в ацетофенон и водород
- гидрирование ацетофенона до этилбензола и воды
Реакции последовательного разложения гидропероксида в условиях окисления вносят довольно заметный вклад в образование побочных продуктов. Чтобы избежать последовательных превращений гидропероксида, ограничивают конверсию этилбензола (до 10 %). Кроме того, для повышения селективности образования гидропероксида этилбензола реакторная система должна быть максимально приближена по гидродинамическому режиму к системе идеального вытеснения (по жидкой фазе). На практике окисление осуществляют в каскаде последовательных реакторов-колонн (обычно больше трех). Для получения алкилароматических гидропероксидов также используют реакторы типа тарельчатой колонны с встроенными змеевиками на каждой тарелке или пустотелой колонны также с встроенными змеевиками.
В кубах ректификационных колонн концентрация стирола повышается до 80-90 % и, хотя присутствие ингибиторов сильно замедляет протекание побочной реакции полимеризации стирола, следует, однако, отметить, что полностью исключить этот процесс не представляется возможным. Поэтому потери стирола будут вследствие его высокотемпературной полимеризации, инициированной полимеризации и сополимеризации.
Высокотемпературная полимеризация - первичный процесс, приводящий к образованию полимера линейного строения. Эта реакция сильно замедляется ингибиторами.
Инициированная полимеризация возникает при попадании пероксидных соединений в жидкость (печное масло), образующуюся после конденсации парообразных углеводородов, отделенных от газов (метана, этилена, водорода и диоксида углерода).
Сополимеризация протекает при наличии в печном масле дивинилбензола, фенилацетилена и других мономеров, что приводит к образованию полимеров разветвленного или сетчатого строения.
Кроме того, при ректификации возможны реакции каталитической пластификации полимера тяжелыми углеводородами, образовавшимися в процессе дегидрирования, особенно при длительном времени пребывания в колоннах. Так, например, нафталин и фенантрен способны реагировать с полимером стирола, разрывая длинные цепи по реакции
Подобные вторичные реакции уменьшают среднюю молекулярную массу полимера, повышают его полидисперсность.
Так и за счет разложения не стойких пероксидных соединений, образующихся при контакте стирола с воздухом уже при комнатной температуре [28, 30, 32]
Для подавления самопроизвольной полимеризации стирола на стадии его выделения из продуктов дегидрирования применяют различные ингибиторы. Способность йода, серы и других веществ замедлять или вообще препятствовать протеканию полимеризации стирола известна давно [33, 34]. Для подавления самопроизвольной полимеризации стирола предлагается широкий класс соединений: моно- и диалкилкатехины, N-алкил-N-арилгидроксиламин, N-циклогексилгидроксиламин, диалкилоксиаминовые соли, монооксим п-хинона, семикарбазиды и их производные, печная сажа в сочетании с органическими пероксидами, N,N-ди-(низший алкид)-гидроксиламиновые соли алифатических карбоновых кислот, трет-аминонафтолы, медные соли ди-2(2-этилгексил)-дитиофосфорной кислоты, диоксим п-хинона, инертные газы [35], ацетаты гидроксиламмония [36], органические эфиры фосфорной кислоты [37], смесь диоксим п-хинона с гидрохиноном [38], бис-(о-изопропилтиокарбонил-гидразон)-N-нафтохинона (БИКН) [39], N,N,N',N'-тетра-(3,5-ди-трет-бутил-4-окси-бензил)-этилендиамин [40].
В промышленности применение получили только гидрохинон, монооксим-п-хинона, п-нитрофенол, смесь диоксим-п-хинона с гидрохиноном [38], сера и смесь серы с трет-бутилпирокатехином [41]. Однако использование указанных ингибиторов не исключает при выделении стирола из печного масла ректификацией образования полимера, накапливающегося в КОРС наряду с ингибиторами и многими другими органическими веществами, из которых идентифицировано от 25 до 40 соединений [42]. Взаимодействие ингибиторов с мономерным стиролом приводит к образованию побочных веществ, также накапливающихся в кубовых остатках.
Кубовые остатки ректификации стирола получаются двумя путями. При одноступенчатом дегидрировании в старых производствах кубовый остаток ректификационных колонн направляется в отгонные кубы, где из него отгоняется стирол. Процесс периодический, проводится под вакуумом до возможно более полного выделения стирола, в результате получается КОРС с содержанием стирола до 35 мас.%. При двухступенчатом дегидрировании кубовые остатки, образовавшиеся в процессе выделения стирола, направляют в роторно-пленочные испарительные аппараты непрерывного действия. Отгонка также производится под вакуумом до содержания мономерного стирола в КОРС до 25 мас.%.
Количество отходов колеблется в зависимости от срока службы катализатора, состава сырья и применяемых ингибиторов от 20 до 30 кг на одну тонну стирола [26].
Различные способы получения стирола. Для выбранного способа привести принципиальную технологическую схему. Обосновать выбор типа и конструкции реактора. Способы управления селективностью.
1) Термическое декарбоксилирование коричной кислоты проводится при температуре 120-130 О С и атмосферном давлении. Выход стирола составляет около 40%

2) Дегидратация фенилэтилового спирта. Реакция может быть реализована как в газовой, так и в жидкой фазе. Жидкофазная дегидратация фенилэтилового спирта осуществляется в присутствии фосфорной кислоты или бисульфита калия. Дегидратация в паровой фазе проводится над катализаторами: оксидами алюминия, тория или вольфрама. При использовании оксида алюминия выход стирола составляет до 90% от теории.

3) Синтез из ацетофенона. Стирол можно получить по реакции ацетофенона с этиловым спиртом над силикагелем:

Выход составляет около 30%.
4) Получение стирола из галогенэтилбензола:


5) Получение стирола дегидрированием этилбензола.
6) Метод производства из этилбензола через гидропероксид этилбензола с одновременным получением оксида пропилена (халкон-процесс):
 | ||
 | ||
7)Получение стирола метатезисом этилена со стильбеном, полученным окислением толуола:

8) Получение стирола каталитической циклодимеризацией бутадиена:

Все приведенные методы получения стирола (за исключением дегидрирования) многостадийны, используют повышенное давление и высокую температуру, что приводит к усложнению и удорожанию производства. Для некоторых методов используется не очень доступное сырье. Небольшие выходы.
Основным методом промышленного производства стирола является каталитическое дегидрирование этилбензола. Этим методом получают более 90% мирового производства этилбензола. В качестве катализаторов дегидрирования применяются сложные композиции на основе оксидов цинка или железа. Раньше наиболее распространенным был катализатор стирол-контакт на основе ZnO. В последнее время используют, главным образом, железо-оксидные катализаторы, содержащие 55-80% Fe2O3; 2-28% Cr2O3; 15-35% K2CO3 и некоторые оксидные добавки. В частности широко используется катализатор НИИМСК К-24 состава Fe2O3 – 66-70%; K2CO3 – 19-20%; Cr2O3 – 7-8%; ZnO2 – 2,4-3,0%; K2SiO3 – 2,0-2,6%. Значительное содержание K2CO3 в катализаторе обусловлено тем, что он способствует дополнительной саморегенерации катализатора за счет конверсии углеродистых отложений водяным паром. Катализатор работает непрерывно 2 месяца, после чего его регенерируют, выжигая кокс воздухом. Общий срок службы катализатора – 2 года.
Реакционный узел для дегидрирования этилбензола можно выполнять различными способами. Один из вариантов – трубчатый реактор, обогреваемый топочным газом по типу, изображенному на рисунке.

Его достоинство – близкий к изотермическому профиль температуры, что позволяет получать повышенную степень конверсии при хорошей селективности. Однако высокие металлоемкость и капитальные затраты на такой реактор привели к созданию других аппаратов – со сплошным слоем катализатора, не имеющих поверхностей теплообмена (рис.А).

Они работают в адиабатических условиях, и реакционная смесь постепенно охлаждается, причем водяной пар играет здесь и роль аккумулятора тепла, не давая смеси чрезмерно охладиться. При получении стирола в единичном адиабатическом реакторе обычная степень конверсии этилбензола составляет около 40%. Недостатки такого единичного реактора – существенное охлаждение смеси, одновременное смещение равновесия в нежелательную сторону и зависящее от этого снижение скорости и селективности. Степень конверсии нельзя довести до приемлемой величины, т.к. это повышает удельный расход пара. Другие установки (рис.Б) приближают процесс к изотермическому и лучше учитывают особенности равновесия реакции. В такой установке имеются 2 реактора (или два слоя катализатора). Охладившуюся в первом реакторе смесь до подачи во второй реактор нагревают перегретым паром. Реактор на рисунке В имеет два-три кольцевых слоя катализатора, причем в первый слой поступает весь этилбензол, но лишь часть водяного пара. В пространство между слоями катализатора подают дополнительное количество перегретого пара. С его помощью повышается температура смеси и происходит ступенчатое разбавление смеси с удалением ее от равновесного состояния, что способствует росту скорости и селективности реакции.

Совмещенное дегидрирование и окисление метанола. Получаемые и побочные продукты. Условия проведения процесса. Особенности оформления реакционного узла.
Дегидрированием или окислением первичных спиртов получают только формальдегид (из метанола). Формальдегид НСНО представляет собой в безводном состоянии бесцветный газ с острым раздражающим запахом (т. конд. -19ОС при 0,1 МПа). При хранении он легко полимеризуется и нередко выпускается в виде твердого полимера — параформальдегида (параформ), который легко деполимеризуется.
Большей частью формальдегид выпускают в виде 37%-ного водного раствора, называемого формалином. В нем формальдегид присутствует в виде гидрата НСНО•Н20 и низкомолекулярньтх полимеров (полиоксиметиленгликоли). Во избежание более глубокой полимеризации и выпадения осадка добавляют к формалину 7—12% (масс.) метилового спирта в качестве стабилизатора.
Совмещенное дегидрирование и окисление метанола. Дегидрирование первичных спиртов, в том числе метанола, менее благоприятно по сравнению с вторичными спиртами по условиям равновесия и селективности реакции. По этой причине, а также с целью устранения эндотермичности процесса осуществили совмещенное дегидрирование и окисление метанола:


Можно так подобрать соотношение этих реакций, чтобы суммарный тепловой эффект был только немного положительным, но достаточным для возмещения потерь тепла в окружающую среду и для нагревания исходной смеси до нужной температуры. Практически при получении формальдегида такое положение достигается, когда процесс на 55% идет через окисление и на 45% через дегидрирование, и тогда процесс можно осуществить в адиабатических реакторах, не имеющих поверхностей теплообмена. В этом состоит одно из преимуществ совмещенного процесса окисления и дегидрирования спиртов. При указанном соотношении реакций дегидрирования и окисления исходная паро-воздушная смесь должна содержать 45% (об.) метанола, что находится за верхним пределом взрываемости метанола в воздухе [34,7% (об.)].
При получении формальдегида кроме основных реакций протекают побочные процессы более глубокого окисления, дегидрирования и гидрирования, ведущие к образованию оксидов углерода, муравьиной кислоты, воды и метана:
Окислительное дегидрирование проводят при недостатке кислорода, поэтому глубокое окисление не получает значительного развития. В то же время само дегидрирование, инициируемое кислородом, протекает быстрее, и все ранее упомянутые побочные реакции не так заметны, как при дегидрировании первичных спиртов. Это позволяет работать при более высокой температуре (500—б00°С), большой скорости реакции и времени контакта 0,01—0,03 с. Выход формальдегида на пропущенное сырье достигает 80-85% при степени конверсии метанола 85-90%. Замечено, что добавление воды к исходному метанолу повышает выход и степень конверсии, по-видимому, в результате разложения ацеталей. Катализаторами синтеза формальдегида этим методом служит металлическая медь (в виде сетки или стружек) или серебро, осажденное на пемзе. Последний катализатор оказался более эффективным и широко применяется в промышленности.
Технологическая схема производства формальдегида окислительным дегидрированием метанола изображена на рис. 139. Метанол, содержащий 10-12% воды, из напорного бака 1 непрерывно поступает в испаритель 2. Туда же через распределительное устройство подают воздух, очищенный от пыли и других загрязнений. Воздух барботирует через слой водного метанола в нижней части испарителя и насыщается его парами. В 1 л образующейся паро-воздушной смеси должно содержаться 0,5 г метанола. Поддержание такого состава смеси очень важно для обеспечения взрывобезопасности и нормального протекания процесса. Поэтому работа испарительной системы полностью автоматизирована: поддерживают постоянные уровень жидкости в испарителе, ее температуру (48—50°С) и скорость подачи воздуха, благодаря чему обеспечиваются необходимые температурный режим и степень конверсии в адиабатическом реакторе.
Паро-воздушная смесь проходит брызгоуловитель, находящийся в верхней части испарителя, затем перегреватель З и поступает в реактор 4, в средней части которого находится катализатор. Реакционные газы сразу же попадают в подконтактный холодильпик 5 (смонтирован вместе с реактором), где происходит быстрое охлаждение смеси и предотвращается распад формальдегида. В разных схемах охлаждение осуществляют проточной водой или паровым конденсатом, когда холодильник играет роль генератора пара низкого, среднего или даже высокого давления. Полученный пар (или горячая вода) служит для перегрева постулающей смеси в теплообменнике 3 и для обогрева испарителя 2.
Охлажденные реакционные газы поступают в абсорбер 6, выполненный в виде тарельчатой колонны; жидкость на тарелках охлаждают внутренними или выносными холодильниками (на схеме не изображены). Абсорбер орошают таким количеством воды, чтобы в кубе получился 36—37%-ный формалина. Стадии абсорбции и разделения продуктов оформляют двумя разными способами.
По одному из них в абсорбере поглощают как форальдегид, так и непревращенный метанол, который содержится в продуктах реакции в количестве, как раз достаточном для стабилизации формальдегида. В этом случае верхнюю тарелку абсорбера охлаждают рассолом, а колонна 7 служит лишь для санитарной очистки газа, в то время как для получения безметанольного формалина (требуемого иногда для ряда целей) необходима установка для отгонки метанола. При втором способе в абсорбере поглощают преимущественно формальдегид; тогда колонна 7 служит для абсорбции метанола, который отгоняют от воды и возвращают на реакцию. В обоих случаях формалин из куба абсорбера б охлаждают в холодильнике 8 и собирают в сборнике 9.

Значительная эндотермичность дегидрирования обусловливает применение трубчатых реакторов, в межтрубном пространств которых циркулируют горячие газы от сжигания газообразного или жидкого топлива. Схема типичного реакционного узла дл дегидрирования спиртов представлена ниже. В топке З происходит сгорание топливного газа, подаваемого вместе с воздухом через специальные форсунки. Температура топочных газов слишком высока, поэтому их разбавляют обратным газом (циркуляция его в системе осуществляется газодувкой 4). Спирт поступает вначале в систему испарителей-перегревателей 1, где он нагревается до нужной температуры частично охлажденными топочными газами. Затем пары спирта попадают в реактор 2, где в трубе находится катализатор. Реакционная смесь подогревается горючими топочными газами, находящимися в межтрубном пространстве, что компенсирует поглощение тепла из-за эндотермичности процесса. По выходе из контактного аппарата реакционные га охлаждают в холодильнике-конденсаторе (на рисунке не показан), а в случае летучих продуктов их дополнительно улавливают водой. Полученный конденсат (и водные растворы) ректифицируют, выделяя целевой продукт и непрореагировавший спирт, возвращаемый на дегидрирование.
Читайте также: