
Полицитемия передается ли по наследству
Обновлено: 10.05.2024
Наиболее часто истинная полицитемия завершается развитием миелофиброза с миелоидной метаплазией (МММ) селезенки. Частота этого исхода составляет 15—20 %, но у проживших более 20 лет она увеличивается до 30 % и более. МММ завершает эритремическую стадию истинной полицитемии. Это естественная эволюция заболевания для всех больных, если они не умирают от тромбозов сосудов, острого лейкоза, других неоплазий, сопутствующих заболеваний и висцеральных осложнений. Продолжительность эритремической стадии в среднем составляет 10—15 лет, но не являются редкостью и более короткие, и более продолжительные ее сроки (в отдельных собственных наблюдениях до 20—25 лет).
Патоморфодинамика этого исхода хорошо изучена с помощью прижизненной трепанобиопсии подвздошной кости и пункции селезенки, патофизиология — с помощью радиологических методов исследования.
Миелоидная метаплазия селезенки (ММС) развивается еще в эритремической (2Б) стадии заболевания и со временем приводит к значительной сплено-мегалии. Обычно она носит трехростковый характер с преобладанием эритропоэза. ММС по времени возникновения опережает развитие миелофиброза.
Развитию коллагенового, определяемого при окраске гематоксилин-эозином миелофиброза предшествует ретикулиновый, для обнаружения которого используются методы импрегнации серебром по Футу и Гомори. На стадии ретикулинового миелофиброза клеточный костный мозг еще весьма гиперплазирован. Коллагеновый миелофиброз вначале сосуществует с клеточной пролиферацией, а потом подавляет ее. Дополнительное развитие остеомиелосклероза наблюдается реже, чем при ХИМФ. Патоморфологическая картина этого периода напоминает изменения костного мозга, наблюдаемые при ХИМФ в 3—4-й стадии его развития.
Это относится и к резко измененному состоянию микро- и макрососудов, мелких артерий и артериол, синусов и синусоидов стромы костного мозга.
При миелофиброзе с миелоидной метаплазией (МММ), развивающемся после истинной полицитемии, значительно меняется топография гемопоэза, что имеет диагностическое значение. Наблюдается падение захвата коллоидного 99mТс осевым скелетом и усиление захвата изотопа эпифизами трубчатых костей, селезенкой и печенью. Возможны различные варианты в степени этих изменений.
Развитие постэритремического миелофиброза происходит под влиянием фиброгенных цитокинов, главным источником которых являются патологические мегакариоциты, тромбоциты и моноциты.
Общность происхождения постполицитемического и ХИМФ отражают и данные цитогенетического исследования.

По данным A. Tefferi и соавт., частота нарушений кариотипа составила 62 % на момент первого исследования, 73 % в динамике заболевания, 90—100 % в период развития острого лейкоза. Характер выявленной цитогенетическои патологии при постполицитемическом МММ был таким же, как и при ХИМФ: 13q-, 20q-, +8, +9. В динамике любого МММ частота 13q- и 20q- значительно возрастает.
Авторы пришли к выводу, что 13q-, 20q- и +8 цитогенетическая патология не имеет плохого прогностического значения. Появление другой цитогенетическои патологии и новых субклонов обычно индуцировано предшествующей цитостатической терапией и имеет плохое прогностическое значение: определены связь между частотой развития острого лейкоза при МММ и обнаружением 5q- в 50 %, 7q- - в 30 %, 12р- — в 25 %, нарушения в хромосоме 1 — в 25 %. Случаев развития острого лейкоза у имевших 13q- не обнаружено.
Клиническая и гематологическая динамика процесса развития миелофиброза с миелоидной метаплазией (МММ) характеризуется прогрессирующим увеличением селезенки, а часто и печени (в единичных случаях гепатомегалия выходит на передний план), потерей массы тела вплоть до кахексии, нередко — появлением субфебрилитета и более высоких подъемов температуры телы у отдельных больных, повышением частоты инфекционных осложнений, проявлениями уратового диатеза, портальной гипертонии, сохранением склонности к тромбозам сосудов и экхимозам.
Большая величина селезенки определяет симптом компрессии желудка и дисфункцию кишечника, смещение левой почки. Возникновение острых болей в области селезенки определяется инфарктами селезенки с возможным развитием периспленита.
Показатели красной крови в процессе развития миелофиброза с миелоидной метаплазией (МММ) постепенно нормализуются, а затем возникает анемия с морфологическими изменениями в эритроцитах, присущими ХИМФ: пойкилоцитоз с грушевидной формой эритроцитов, анизоцитоз и нормобластемия. Характерно нарастание числа лейкоцитов и левого сдвига в лейкоцитарной формуле крови; у отдельных больных развивается лейкопения. Число тромбоцитов варьирует от высоких до низких значений.
На поздних этапах миелофиброза с миелоидной метаплазией (МММ) нередко возникает макроцитарная анемия с токсигенной зернистостью в эритроцитах, шизоцитами и кольцами Кэбота. Причиной ее развития является относительный дефицит фолиевой кислоты, а не витамина В12.
Основные причины развития анемии при постполицитемическом МММ — повышенный гемолиз эритроцитов в резко увеличенной селезенке, неэффективный эритропоэз с гибелью части эритроидных предшественников и коммитированных клеток в костном мозге, доказанной радиологическими исследованиями, подавление эритропоэза диффузным миелофиброзом и лейкемизированным миелопоэзом. К ним добавляются такие факторы, как гидремия, дефицит железа, оставшийся от эритремической стадии, относительный дефицит фолиевой кислоты. Отдельные причины часто сочетаются у одного и того же больного.
В зависимости от причин развития анемический синдром имеет различную тяжесть и прогноз от вполне благоприятного при железодефицитной до тяжелого при рефрактерной анемии и миелодисплазии, одним из вариантов которой является сидеробластная анемия.
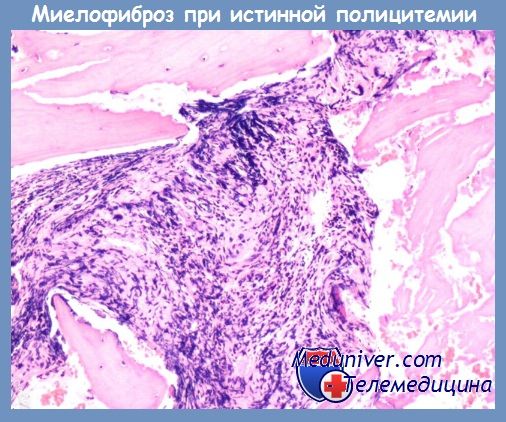
Течение постэритремической миелоидной метаплазии селезенки и миелофиброза необычайно вариабельно. У некоторых больных оно вполне доброкачественное с медленными темпами роста размеров селезенки и печени, нормальными показателями красной крови, умеренным лейкоцитозом и тромбоцитозом или без них. Подобную многолетнюю благополучную динамику заболевания можно обозначить как период стабилизации. У других больных отмечается быстрое прогрессирование спленомегалии, анемический синдром, нарастающий лейкоцитоз с малопроцентной бластемией.
Эти варианты лейкоцитоза, зрелоклеточного или напоминающего хронический миелолейкоз, с высокой вероятностью предвещают развитие миелодисплазии или острого лейкоза. Это стандартный исход истинной полицитемии, протекающей с лейкоцитозом выше 30•109/л.
Плохое прогностическое значение имеет и лейкопения меньше 3•10 9 /л, особенно если она сочетается с выраженной анемией и омоложением лейкоцитарной формулы. Подобного рода состояние, как и лейкоцитозные формы при наличии рефрактерной к лечению анемии, сопровождающееся повышением температуры тела и/или появлением геморрагического синдрома, рассматривается как миелодиспла-зия, предстадия острого лейкоза. Так же трактуются и случаи сидеробластной анемии. В этих случаях можно ожидать прогностически неблагоприятных цитогенетических находок.
Итальянские авторы рассматривают всю стадию постполицитемического МММ как предостролейкозную с вероятностью развития острого лейкоза через 3 года после ее диагностики. Если с первым положением условно можно согласиться, то это не относится к обозначенному авторами сроку, поскольку у многих больных он достаточно высокий, а часть больных и не доживает до его развития.
Манифестации острого лейкоза предшествует помимо рефрактерной анемии и нарастающего лейкоцитоза ряд признаков:
• асептическая лихорадка, продолжительность которой до диагностики острого лейкоза может составить 1—2 года;
• глубокая цитопения, в том числе и тромбоцитопения, немотивированная большой величиной селезенки и предшествующей цитостатической терапией;
• быстрый рост размеров селезенки, особенно если он сочетается с лихорадочным синдромом.
• тромбоцитемия на позднем этапе заболевания, если ее раньше не было (2 собственных наблюдения).
Манифестации острого лейкоза могут предшествовать трудно квалифицируемые дерматиты лица, глосситы, резистентность к прежде адекватному лечению. Подозрение на острый лейкоз возникает и в случаях захвата осевым скелетом 99mТс при наличии выраженного миелофиброза в гистоморфологическом препарате.
Развитие острого лейкоза возможно как в эритремической стадии заболевания, так и в стадии постэритремической ММС и миелофиброза. В последнем случае преобладают лейкоцитозные варианты с частичным вызреванием, в крови встречаются эритрокариоциты, осколки ядер мегакариоцитов. Чаще всего имеет место миелобластный вариант острого лейкоза, но возможны и наблюдались нами эритромиелоз, миеломоноцитарный и лимфобластный варианты. Для клинических проявлений острого лейкоза во всех случаях были характерны мучительные оссалгии.
К настоящему времени уже нет сомнений в том, что естественные тенденции к завершению заболевания острым лейкозом при истинной полицитемии имеются, но они выражены незначительно. У леченных цитостатическими препаратами частота развития острого лейкоза существенно возрастает, как и заболеваемость раком и злокачественными лимфомами.
По мере получения доказательств лейкозогенности цитостатической терапии стимулировались поиски новых методов лечения.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021

Что собой представляет эритремия, или истинная полицитемия? Если описать эту болезнь простейшими словами, эритремия означает, что организм человека вырабатывает слишком много красных кровяных телец. Современная наука установила принадлежность данной патологии к группе под названием миелопролиферативные заболевания, или миелопролиферативные опухоли. Патологии этой группы поражают костный мозг. Развивается хроническая лейкемия, приводящая к избыточной выработке клеток костным мозгом. Если у вас эритремия, это означает чрезмерное количество красных кровяных телец, но в некоторых случаях в избытке производятся белые тельца или тромбоциты.
Отчего развивается эритремия?
Очевидно, что мутация Янус-киназы 2 не является единственной причиной возникновения описанной патологии. У 95% пациентов с эритремией наблюдается данное генное изменение, однако у других больных обнаруживаются иные мутации генов, которые, предположительно, оказывают на клетки аналогичное влияние.
Как проводится диагностика эритремии в Израиле?
Первыми симптомами эритремии обычно являются:
- Повышенная усталость;
- Сильный зуд после принятия ванны;
- Сыпь.
Возможны и другие симптомы, так как заболевание может проявляться по-разному.
В дополнение к общему анализу крови, а также проверке на обнаружение мутации Янус-киназы 2, врачи обычно назначают биопсию костного мозга. Заболевание поражает костный мозг, поэтому исследование изнутри позволяет выявить как непосредственно патологию, так и стадию ее развития.
Программа диагностики эритремии в Топ Ихилов
День 1-й. Лабораторные исследования
Пациент сдает анализы крови:
- Общий;
- Биохимический;
- Мазок периферической крови.
День 2-й. Прием гематолога
Пациента принимает ведущий гематолог клиники Топ Ихилов – доктор Фреди Авив. Он сообщает больному его точный диагноз, включая стадию заболевания, назначает лечение и отвечает на все вопросы пациента.
Стоимость программы обследования – $1123 . Диагностика занимает 2-3 дня.
✓ Прямо сейчас – Возможность получить бесплатную консультацию врача Топ Ихилов
Важно верно определить тип болезни, прежде чем начинать лечение по месту жительства. Прямо сейчас бесплатно проконсультируйтесь с израильским врачом. Узнайте, сколько стоят в Топ Ихилов нужные вам диагностические процедуры.
Запросить цену диагностики в Ихилов
Как развивается заболевание?
Эритремия – это болезнь, способная по-разному действовать на людей. Как уже было сказано, ее симптомы достаточно многообразны. Со временем она может привести к таким осложнениям, как тромбозы или кровоизлияния. Запускание болезни в течение длительного времени чревато ее прогрессированием. Эритремия не всегда прогрессирует, однако при наличии данного нарушения у пациента на протяжении 10 лет и более возникает риск обострения. Обострение патологии может протекать по одному из нескольких разных сценариев.
Второй сценарий обострения эритремии – это ее преобразование в процесс, который мы называем «острой лейкемией«. И это очень тяжелое заболевание.
Отзыв о лечении лимфомы в Топ Ихилов, Александр, Россия
Чем отличается лечение эритремии в Израиле от лечения в странах СНГ?
- Высокая квалификация израильских гематологов. Чтобы получить право на самостоятельную врачебную практику, гематолог в Израиле обучается в течение 16 лет. Курс обучения включает не только теоретические занятия, но и специализации в ведущих клиниках мира.
- Гарантированное качество лекарственных препаратов. В Топ Ихилов для лечения эритремии применяются циторедуктивные препараты израильского производства, защищенные от подделки. Минздрав Израиля строго контролирует весь процесс производства, хранения и транспортировки лекарств, поэтому их высокое качество гарантировано.
Врачи, занимающиеся диагностикой и лечением эритремии в Топ Ихилов


Получить бесплатную консультацию врача Ихилов
Какими методами лечат эритремию в Израиле
При лечении заболеваний в Израиле необходимо принимать в расчет как краткосрочные, так и долгосрочные перспективы. Что касается краткосрочных перспектив, существуют методы лечения, направленные на снижение риска тромбозов и кровоизлияний. В отношении долгосрочных перспектив, к сожалению, на данный момент не существует терапии, которая однозначно бы уменьшала риск обострения заболевания и возникновения осложнений в виде миелофиброза или острой лейкемии.
Рассмотрим подробнее доступные методы лечения эритремии. Вполне очевидно, что я отправлю вас к вашему врачу, который сможет разработать схему терапии, идеально подходящую именно для вас. В целом, впрочем, известно, что малая доза аспирина – при отсутствии аллергии на аспирин, естественно, – помогает большинству пациентов избежать риска развития тромбоза. Кроме того, снижение количества красных кровяных телец посредством кровопускания, или флеботомии, позволяет поддерживать относительно нормальный гематокрит [1] . Гематокрит – это процентное соотношение кровяных телец и плазмы. Нормальный показатель составляет 45% у мужчин и около 42% у женщин. Пациенты сдают кровь точно так же, как доноры, однако в настоящее время банкам крови запрещено использовать данный биоматериал для переливания другим больным. Тем не менее, таким способом можно контролировать течение заболевания.

И наконец, отдельным пациентам назначаются медикаменты, снижающие число форменных элементов крови до нормального показателя. Кто входит в данную группу? Это те люди, кому с наибольшей вероятностью угрожает тромбоз или кровоизлияние. В первую очередь это касается пациентов старше 62 лет, а также тех, у кого ранее наблюдались тромбозы или кровоизлияния. Третья категория – пациенты с существенным риском развития сердечно-сосудистых заболеваний или значительно повышенным уровнем тромбоцитов (приблизительно свыше 1,5 млн на 1 кубический сантиметр).
В данный момент ученые работают сразу в нескольких направлениях, пытаясь определить, каким именно пациентам с наибольшей вероятностью угрожает тромбоз. Эти сведения позволят максимально усовершенствовать терапию.
Для снижения количества кровяных телец при эритремии назначаются:
Получить программу лечения и точные цены
Доктор Ирина Стефански о целесообразности повторной диагностики в Израиле
Стоимость лечения эритремии в Израиле
| Эритремия: стоимость диагностики и лечения в клиниках Израиля | Цены (USD) |
|---|---|
| Консультация гематолога | 543 |
| Биопсия костного мозга | 156 |
| Общий анализ крови | 75 |
| Кровопускание (флеботомия) | 53 |
Инновации: каким образом терапия эритремии изменится в будущем
У пациентов с миелопролиферативными заболеваниями и в особенности – с эритремией есть многообещающее будущее. Сейчас об этой патологии известно гораздо больше, чем, к примеру, 5 лет назад. Научные исследования ведутся по многим направлениям, разрабатываются новые медикаменты и способы не только облегчить состояние пациентов, не только снизить риски в краткосрочной перспективе, но и, по возможности, повысить контролируемость заболевания в долгосрочной перспективе.
ФГБУ "Гематологический научный центр" Минздрава России, Москва
Научно-клиническое отделение гематологической хирургии Гематологического научного центра Минздрава РФ, Москва
ФГБУ "Гематологический научный центр" Минздрава России, Москва
ФГБУ "Гематологический научный центр" Минздрава России, Москва
Гематологический научный центр Минздрава России, Москва
ГНЦ Минздрава России, Москва
Диагностика латентной истинной полицитемии (взгляд клинициста)



ФГБУ "Гематологический научный центр" Минздрава России, Москва
Научно-клиническое отделение гематологической хирургии Гематологического научного центра Минздрава РФ, Москва
ФГБУ "Гематологический научный центр" Минздрава России, Москва
ФГБУ "Гематологический научный центр" Минздрава России, Москва
Гематологический научный центр Минздрава России, Москва
ГНЦ Минздрава России, Москва
ВОЗ — Всемирная организация здравоохранения
ИП — истинная полицитемия
КМ — костный мозг
МПН — миелопролиферативное новообразование/неоплазия/опухоль
ОЦЭ — объем циркулирующих эритроцитов
ТО — тромботические осложнения
ЭТ — эссенциальная тромбоцитемия
BCSH — British Committee for Standards in Haematology; Британский комитет по стандартизации в гематологии
Истинная полицитемия (ИП), или эритремия, болезнь Вакеза, истинная красная полицитемия — клональное миелопролиферативное новообразование (неоплазия, опухоль) — МПН, которое характеризуется пролиферацией эритроидного, гранулоцитарного, мегакариоцитарного ростков миелопоэза (панмиелоз), с преимущественной пролиферацией эритроидного ростка кроветворения, увеличением количества эритроцитов и гемоглобина, тромбоцитозом, лейкоцитозом в периферической крови (панцитоз), независимостью эритропоэза от нормальных механизмов регуляции. Почти все больные являются носителями мутации JAK2V617 °F или другой функционально сходной мутации [1].
Согласно классификации Всемирной организации здравоохранения (ВОЗ) от 2008 г. диагноз ИП должен быть установлен на основании комплексной оценки клинической картины и лабораторных показателей. Ведущим диагностическим критерием является содержание гемоглобина >165 г/л у женщин и >185 г/л у мужчин [2]. В соответствии с критериями Британского комитета по стандартизации в гематологии (British Committee for Standards in Haematology — BCSH) в случаях с пограничными уровнями гемоглобина диагностика ИП предполагает оценку гематокрита: >52% у мужчин и >48% у женщин [3].
Концентрации гемоглобина и гематокрит служат маркерами, позволяющими косвенно определить объем циркулирующих эритроцитов. Как известно, содержание гемоглобина и гематокрит определяются объемом циркулирующих эритроцитов (ОЦЭ) и объемом циркулирующей плазмы. Определение ОЦЭ позволяет отличить абсолютный эритроцитоз от относительного, причиной которого служит уменьшение объема плазмы. Однако важность определения ОЦЭ в дифференциальной диагностике эритроцитозов, невозможно переоценить. При И.П., в отличие от эритроцитозов, вызванных избыточной продукцией эритропоэтина (ЭПО), объем плазмы часто повышен. Из-за этого содержание гемоглобина и гематокрит возрастают в меньшей степени, чем ОЦЭ. Как у мужчин, так и у женщин повышение концентрация гемоглобина не всегда связано с абсолютным эритроцитозом и, наоборот, низкое содержание гемоглобина может наблюдаться при абсолютном эритроцитозе [4, 5]. Незнание этого приводит к тому, что истинная причина тромбозов печеночных и воротной вен у больных с так называемыми миелопролиферативными неоплазиями неклассифицированными остается неустановленной [6].
Единственным надежным методом дифференциальной диагностики абсолютных и относительных эритроцитозов является определение массы циркулирующих эритроцитов — изотопное разведение с использованием собственных эритроцитов больного, меченных 51 Cr [7]. Однако данная методика не используется в ежедневной клинической практике, так как является трудоемкой, дорогостоящей и имеет недостаточную диагностическую точность.
В классификации ВОЗ представлена следующая модель клинического течения ИП [2].
Первая стадия полицитемии (начальная) характеризуется умеренным эритроцитозом, невысоким содержанием гемоглобина и гематокритом. В ряде случаев ИП может имитировать эссенциальную тромбоцитемию (ЭТ). Селезенка несколько увеличена, но пропальпировать ее обычно не удается (увеличение селезенки обусловлено повышенной секвестрацией в ней тромбоцитов и эритроцитов).
Вторая стадия полицитемии (развернутая, пролиферативная) характеризуется выраженной плеторой, гепато- и спленомегалией. В крови обнаруживают эритроцитоз, тромбоцитоз, лейкоцитоз со сдвигом лейкоцитарной формулы влево.
Третья стадия полицитемии (постполицитемический миелофиброз) характеризуется гепато-, спленомегалией, в них обнаруживают очаги экстанодального кроветворения. В крови нарастает панцитопения, в костном мозге (КМ) — фиброз разной степени выраженности.
Ряд исследователей начальную стадию ИП выделяют как отдельную форму заболевания: замаскированная, в процессе эволюции, скрытая, субклиническая, тлеющая, преполицитемическая, нераспознанная/ недиагностированная И.П. Латентная И.П. характеризуется наличием мутации JAK2V617 °F, морфологическими особенностями КМ, соответствующими критериям ВОЗ от 2008 г. для ИП, содержанием гемоглобина ниже диагностического ( 65 лет и лейкоцитоз >10·10 9 /л были независимыми факторами неблагоприятного прогноза. Авторы не исключают влияние эпигенетических механизмов, определяющих как фенотип заболевания, так и быструю прогрессию заболевания [8].

Таблица 1. Клинико-лабораторные характеристики больных с латентной ИП и развернутой ИП Примечание. ИМ — инфаркт миокарда; ОНМК — острое нарушение мозгового кровообращения.
Морфологическая характеристика трепанобиоптата КМ у больных с латентной ИП не отличалась от таковой у больных с явной ИП.
Дифференциальная диагностика в группе классических Ph-негативных МПН проводится на основе данных анамнеза, клинических и лабораторных характеристик, морфологического исследования трепанобиоптата КМ [11—16]. Большим главным диагностическим критерием ИП, косвенно отражающим массу циркулирующих эритроцитов, согласно классификации ВОЗ от 2008 г., является концентрация гемоглобина и гематокрит. В ряде случаев при морфологических признаках ИП клинические или лабораторные характеристики не соответствуют диагнозу ИП [17]. Определение ЭПО в сыворотке крови может служить только дополнительным диагностическим критерием, поскольку ЭПО ниже нормы или субнормальный выявлен в 60% наблюдений у больных ИП, а нормальные значения найдены в неоспоримых случаях ИП [18, 19].
Выявленное несоответствие между клинико-лабораторными и морфологическими характеристиками послужило поводом для проведения ряда клинических исследований, результатом которых явился пересмотр диагностических критериев ИП. В 2014 г. предложены новые диагностические критерии ИП (табл. 2) [20—22]. Первой и самой значительной поправкой является снижение порогового диагностического уровня гемоглобина. Во-вторых, гематокрит введен как дополнительный критерий для оценки ОЦЭ. При сравнении критериев ВОЗ, основанных на концентрации гемоглобина, и пересмотренных критериев BCSH выявлено, что критерии BCSH более специфичны для диагностики И.П. Предполагается, что гематокрит более специфичен в качестве индикатора повышения ОЦЭ, чем гемоглобин [23]. В-третьих, гистологическое исследование трепанобиоптата КМ имеет ведущее диагностическое значение, а также может играть важную роль в дифференциальной диагностике ИП с другими вариантами МПН и реактивными изменениями [24, 25].

Таблица 2. Диагностические критерии ИП
Таким образом, у ряда пациентов с мутацией JAK2V617 °F, гистологической картиной ИП в трепанобиоптате КМ (расширение трех ростков кроветворения в разных соотношениях — панцитоз) лабораторные признаки, в частности содержание гемоглобина и гематокрит, не соответствуют диагностическим критериям ВОЗ от 2008 г. для И.П. Данная форма заболевания (латентная ИП) по сравнению с явной ИП характеризуется преобладанием среди больных мужчин, более высоким тромбоцитозом, низким количеством лейкоцитов, низкой аллельной нагрузкой JAK2V617 °F. Наши данные соответствуют наблюдениям зарубежных авторов. Преобладание женщин как в группе с латентной, так и явной ИП в нашем исследовании можно объяснить преобладанием числа женщин над мужчинами в общей популяции и тем, что женщины обращаются за медицинской помощью чаще, видимо, в силу более внимательного отношения к своему здоровью.
Статистический анализ факторов риска развития тромбогеморрагических осложнений при латентной ИП и явной ИП не выявил различий [26]. Результаты нашего исследования показали, что у пациентов с латентной ИП риск развития ТО выше, чем при явной ИП: 38 и 16% случаев соответственно. Эти же результаты подтверждают данные F. Lussana и соавт. [10] о преобладании венозного тромбоза над артериальным в группе молодых больных. Следует помнить, что у пожилых людей причиной тромбоза наиболее часто бывает атеросклероз. Молодые пациенты с МПН с пограничным содержанием гемоглобина без сопутствующей сердечно-сосудистой патологии относятся к группе низкого риска развития тромбогеморрагических осложнений. По этой причине нередко выбирается наблюдательная тактика лечения. Согласно данным литературы высокая частота тромбозов у больных латентной ИП может быть обусловлена менее интенсивной терапией. Вследствие высокого риска развития ТО рекомендации по тактике лечения больных с латентной ИП не должны отличаться от рекомендаций для больных с явной ИП, в том числе касающиеся поддержания гематокрита ниже 45% [27, 28].
Согласно данным литературы латентная ИП характеризуется быстрым прогрессированием и развитием постполицитемического миелофиброза и трансформацией в острый лейкоз [11—13]. Однако нельзя исключить, что латентная ИП может иметь более длительный анамнез до установления диагноза по сравнению с И.П. Этой особенностью течения заболевания можно объяснить низкую общую выживаемость больных с латентной ИП [8]. Оценить выживаемость в нашей группе больных не представляется возможным вследствие малого периода наблюдения.
Заключение
Результаты нашего исследования показывают, что МПН с мутацией JAK2V617 °F, пограничным содержанием гемоглобина, морфологическими особенностями трепанобиоптата КМ, характерными для ИП, представляют собой отдельный вариант ИП — латентную И.П. Еще раз необходимо подчеркнуть, что выделение латентной ИП как отдельного нозологического варианта имеет большое клиническое значение, поскольку, несмотря на несоответствие всем критериям ВОЗ от 2008 г., больные имеют высокий риск развития тромбогеморрагических осложнений, как и больные с явной ИП.
Хронические миелопролиферативные заболевания — это группа патологий, при которых происходит неконтролируемый рост кровяных клеток. Нарушение вызвано генетическими мутациями.
Акции

Запись на консультацию со скидкой 10%.



Консультация врача-хирурга по поводу операции бесплатно!
- Врачи
- Цены
- Лимфома
- Хронические миелопролиферативные заболевания
- Множественная миелома
- Лимфома Ходжкина
Содержание статьи:
ХМПЗ (хроническое миелопролиферативное заболевание) — это группа патологических состояний организма, течение которых характеризуется неконтролируемым ростом клеток крови. Данное нарушение возникает в результате генетических мутаций. К ХМПЗ относят эозинофильный и нейтрофильный рак крови, миелолейкоз, эритремию, сублейкемический миелоз и хроническое миелопролиферативное заболевание эссенциальная тромбоцитемия.
Распространенность ХМПЗ невысокая. Обычно заболевает не более 1 человека на 100 000 населения. Точные причины развития данной патологии неизвестны. В ряде случаев имеется связь с наличием в анамнезе пациента других онкологических заболеваний, при лечении которых использовалась лучевая терапия и цитостатики.
Чаще всего заболевание диагностируют у людей среднего (от 40 лет) и пожилого возраста. У всех видов ХМПЗ есть общие характеристики, а именно:
- происхождение из одной стволовой кроветворной клетки;
- в зависимости от уровня поражения пролиферируют один либо несколько рядов клеток;
- наблюдается перепроизводство кровяных клеток без существенной дисплазии;
- развитие миелофиброза и экстрамедуллярного кроветворения;
- развитие острого лейкоза.
Эффективность терапии хронической миелопролиферативной болезни зависит от вида патологии и ее стадии.
Классификация хронических миелопролиферативных заболеваний
Выделяют следующие виды ХМПЗ:
- хронический миелолейкоз (ХМЛ);
- нейтрофильный лейкоз;
- эозинофильный лейкоз;
- ХМПЗ истинная полицитемия;
- эссенциальная тромбоцитемия;
- идиопатический миелофиброз;
- не классифицируемое ХМПЗ (когда развитие и клинические признаки не соответствуют ни одной из вышеперечисленных форм заболевания).
Все ХМПЗ характеризуется отсутствием диспластических изменений в костном мозге и кровяных клетках на начальных этапах заболевания.
Помимо вышеперечисленных видов в современной онкологии отдельно выделяют группу с миелоидной дисплазией, которая занимает промежуточное положение между ХМПЗ и миелодиспластическим синдромом.
Классификация ХМПЗ с миелодисплазией:
- миеломоноцитарный хронический лейкоз;
- миеломоноцитарный хронический ювенильный лейкоз;
- хронический атипичный миелолейкоз;
- не классифицируемое ХМПЗ с миелодисплазией (когда развитие и клинические признаки не соответствуют ни одной из вышеперечисленных форм заболевания).
Причины и факторы риска развития ХМПЗ
МПЗ относятся к группе клональных заболеваний. Патологические изменения в организме при таких болезнях начинаются с одного либо нескольких сбоев в ДНК всего лишь одной стволовой клетки костного мозга.
Недифференцированная (гемопоэтическая) стволовая клетка - это незрелая кровяная клетка, которая в итоге может превратиться в один из элементов крови (лейкоцит, эритроцит, тромбоцит). В результате патологических изменений в ДНК этой клетки она начинает очень быстро делиться, что приводит к образованию большого количества патологических стволовых клеток, которые созревая, превращаются в один из вышеперечисленных элементов крови.
По мере накопления патологически измененных клеток крови состояние пациента начинает ухудшаться. В большинстве случаев причина, по которой произошел запуск патологического процесса в костном мозге, остается неизвестной.

Одним из основных факторов риска при миелопролиферативных патологиях является возраст больного. После 70 лет увеличивается клональное кроветворение, что повышает риск развития ХМПЗ в 11-13 раз.
Симптомы
Клинические проявления хронических миелопролиферативных заболеваний крови не специфичны, поэтому необходима тщательная дифференциальная диагностика с другими патологиями. На начальном этапе, когда опухолевая масса небольшая, у пациентов наблюдается умеренный лейкоцитоз.
Клинически заболевание может проявляться следующими симптомами:
- хроническая усталость;
- беспричинная слабость, повышенная утомляемость;
- тяжесть в подреберье;
- беспричинная потеря веса;
- повышенное потоотделение;
- дискомфорт в желудке;
- периодические расстройства кишечника;
- образование гематом;
- периодическая асфиксия;
- отеки конечностей;
- постоянная субфебрильная температура тела;
- боли в суставах;
- слабо болезненная, увеличенная селезенка, печень;
- нарушения слуха;
- периодические обмороки;
- изменение окраски кожи.
При истинной полицитемии у пациентов повышается уровень гемоглобина в крови, что проявляется сильным покраснением кожи лица, которое многие ошибочно принимают за проявление алкоголизма. Кроме этого, одним из характерных симптомов данного заболевания ХМПЗ является кожный зуд, возникающий при контакте с водой. Одним из самых опасных состояний при ХМПЗ является тромбоцитоз (склонность к образованию тромбов). Это резко повышает риск закупорки крупных сосудов, что приводит к развитию инсультов, инфарктов и угрожает не только здоровью, но и жизни пациента.
Течение ХМПЗ хроническое. При отсутствии лечения болезнь со временем прогрессирует с возможным развитием острого миелолейкоза, миелодиспластического синдрома. Кроме этого наблюдается прогрессирующее увеличение размеров селезенки.
Диагностика хронической миелопролиферативной болезни
Основными методами диагностики ХМПЗ являются лабораторные методы исследования и биопсия.
Лабораторные методики
- Анализ крови (общий) с определением лейкоцитарной формулы.
- Микроскопическое исследование мазка крови.
- Выявление уровня щелочной фосфотазы.
- Анализ крови на маркеры ХМПЗ:
- FISH или ПЦР периферической крови — проводится для выявления мутации bcr-abl, что помогает диагностировать хронический миелолейкоз и дифференцировать его от других видов ХМПЗ;
- ПЦР на определение мутации JAK2 — позволяет выявить эссенциальную тромбоцитопению, истинную полицитемию и миелофиброз.
Биопсия
Взятие образцов костного мозга и последующее их исследование под микроскопом проводятся в большинстве случаев. При помощи данных методик проводят дифференциальную диагностику ХМПЗ от миелодиспластического синдрома. При проведении гистологии образцов костного мозга обнаруживают повышенное содержание паренхиматозных клеток. Миелофиброз диагностируют путем окрашивания образцов ткани ретикулином.
Пациенты с установленными диагнозами остаются под наблюдением гематолога пожизненно.
Методы лечения ХМПЗ
Для лечения хронических миелопролиферативных заболеваний часто применяют цитостатики. Препараты из этой группы обладают способностью воздействовать на конкретный гематологический росток, что позволяет уменьшить скорость патологического деления клеток крови.
Кроме этого, для лечения ХМПЗ могут использоваться препараты, которые снижают негативные последствия полицитемии (глюкокортикостероиды, средства для разжижения крови, противотромботические соединения и ряд других). С их помощью можно убрать повышенную вязкость крови, которая является следствием усиленного размножения кровяных клеток, снять общее воспаление, предотвратить тромбообразование.
![07_treatment]()
Крайне важно при терапии ХМПЗ остановить аномальное увеличение селезенки. Для этого используют лучевую терапию. Если лечение не дает эффекта, то проводят операцию по удалению селезенки.
Кроме этого, для каждого вида ХМПЗ существуют свои специфические методы терапии. Например, при эозинофильном лейкозе показан прием антигистаминных средств, которые снижают проявления аллергии. При истинной полицитемии одним из вариантов терапии является кровопускание. При тромбоцитемии больным назначают антикоагулянты.
Если ХМПЗ диагностируют у молодых людей (что бывает редко), то возможно проведение радикального лечения, которое включает в себя пересадку стволовых клеток и приводит к полному выздоровлению. К сожалению, большинство пожилых людей в силу возраста не могут перенести эту операцию.
Прогноз при ХМПЗ крови
![08_forecast]()
При хроническом миелопролиферативном лейкозе в течение 3-5 лет идет хроническое развитие заболевания. Затем наступает акселерированная фаза, которая переходит в бластный криз. После этого прогноз выживаемости — от нескольких месяцев до года.
Выживаемость при хронических миелопролиферативных болезнях при грамотной своевременно начатой терапии составляет около 20 лет. Если лечение не проводится, то пациент умирает в течение полутора лет. При этом с каждым месяцем повышается риск развития тромбоэмболий, что еще более ухудшает прогноз. Запущенная стадия заболеваний проявляется снижением уровня эритроцитов, что сопровождается усилением спленомегалии и изменениями в костном мозге.
Профилактика ХМПЗ
Эффективной специфической профилактики не существует. Для снижения вероятности развития ХМПЗ необходимо соблюдать следующие рекомендации:
- правильно питаться;
- отказаться от вредных привычек;
- соблюдать режим труда и отдыха;
- больше двигаться, заниматься спортом;
- избегать работы на вредных производствах, особенно там, где организм подвергается воздействию ионизирующего излучения;
- не реже раза в год посещать врача для проведения комплексного профилактического обследования организма, это поможет выявить возможные патологи на ранних стадиях, что существенно облегчит последующее лечение и улучшит прогноз.
Информация в статье предоставлена в справочных целях и не заменяет консультации квалифицированного специалиста. Не занимайтесь самолечением! При первых признаках заболевания необходимо обратиться к врачу.
Читайте также:
- Как рассчитать выходное пособие при суммированном учете рабочего времени
- Временная нетрудоспособность с 61 дня сбербанк что это значит
- Как называется обработка и сведение в одну систему всего законодательного материала
- Банк обвиняет в мошенничестве что делать
- Национальное и международное право что важнее эссе

