
Передается ли синдром гольденхара по наследству
Обновлено: 02.07.2024
Наследственные факторы
По данным многих исследований существуют структурные, анатомические и генетические взаимосвязи между такими патологическими состояниями, как микротия, суженные и оттопыренные уши. Показано, что эти деформации взаимосвязаны и могут быть наследственными.
Выявлены как доминантные, так и рецессивные факторы наследования при глухоте, связанной с патологией ушных раковин. Деформации ушных раковин часто встречаются в семьях с синдромом Тричер-Коллинза (мандибулофациальный синостоз).
В исследовании семей с микротией у одного или обоих родителей с исключением хромосомных аберраций пришли к выводу, что наследование должно быть мультифакторным и что риск рецидива составляет в среднем 5,7% (от 3 до 8%) [95]. Среди ближайших родственников помимо различных дефектов и деформаций ушной раковины наблюдалось нарушение развития челюстей и лицевого нерва.
Специфические факторы
Среди ученых есть предположение, что ишемия тканей (снижение кровотока) вследствие закупорки артерии может стать причиной патологического развития ушных раковин. Это говорит о том, что причиной развития деформации чаще являются проблемы, возникающие во время развития плода, а не наследственные факторы. В поддержку этой теории выступает факт частого наличия микротии только у одного из близнецов. Детские дизморфологи заявляют, что многие виды врожденных уродств чаще встречаются при многоплодной беременности, и что это может быть вследствие явления под названием синдром "фетоплацентарного обкрадывания", когда плацента нормального двойника часто больше, чем плацента аномального, и, возможно, происходит усугубление кровообращения одного из близнецов, развитие которого затем нарушается.
Хорошо известно, что появление глухоты и иногда микротии происходит в результате краснухи в течение первого триместра беременности. Кроме того, использование некоторых лекарств в этот критический период тоже может стать причиной врожденных заболеваний ( например, талидомида, роаккутан (Accutane), кломид, ретиноевая кислота).
Влияние различных внешних факторов во время первого триместра беременности на развитие микротии у ребенка не однозначно, в 1-4% случаев это могут быть простудные заболевания, корь, травмы, облучение, месячные во время первого триместра, гиперемезис (чрезмерная рвота), наличие диабета, эмоциональные перегрузки, употребление алкоголя, лекарства от тошноты.
В заключение, развитие микротии – это, как правило, случайное спорадическое событие, и важно, чтобы родители понимали, что деформация не была вызвана какими-либо действиями матери до или во время беременности, и что риск рецидива в семье составляет около пяти процентов, или, иначе говоря, один из двадцати.
Ассоциированные патологии
Эмбриологическое развитие подразумевает, что микротия обычно сопровождается патологией среднего уха. При классической микротии обычно присутствует атрезия слухового канала и нарушения слуховых косточек - Синдром Конигсмарка (микротия, атрезия наружного слухового прохода и кондуктивная тугоухость).
Деформация среднего уха может варьироваться от суженного канала и незначительных нарушений слуховых косточек до расплавленных гипоплазированных слуховых косточек и недостаточной аэрации сосцевидной кости. Следует обратить внимание, что, поскольку у пациентов с атрезией есть слуховые трубы, как и у всех, они могут спровоцировать воспаление среднего уха (средний отит) даже если они не имеют наружный слуховой проход. Поэтому, если этот диагноз невозможно подтвердить с помощью отоскопии, при наличии подозрения на средний отит в деформированном ухе, разумно назначить антибиотики.
Патология челюстно-лицевой области
Так как ушная раковина развивается из тканей жаберных дуг, не удивительно, что у значительного процента пациентов с микротией обнаруживают дефицитные компоненты лица, которые происходят из этих эмбриональных элементов. Проявление в виде уменьшенной половины лица, состояние, известное как гемифациальная микросомия – это, в основе, недоразвитие челюстей и вышележащих мягких тканей. Наиболее полное генетическое проявление этого состояния включает в себя дефекты наружного и среднего уха, гипоплазию верхней и нижней челюстей, скуловой и височной костей, макростому и боковые щели лица, парез лицевого нерва, атрофии мышц лица и околоушной железы, даже небные мышцы могут быть ослаблены на вовлеченной стороне.
Почки и мочевыводящие пути
Нарушения урогенитального тракта увеличиваются при наличии деформаций ушной раковины, особенно когда пациент страдает от других проявлений недоразвития лица. У некоторых пациентов может наблюдаться недоразвитие половых органов (гипоспадия, агенезия женских половых органов), различные нарушения почек (подковообразная почка, односторонняя почечная агенезия, тазовое расположение почки и т.п.). Однако эти нарушения не вызывают жизненно-опасных нарушений работы органов мочеполовой системы. Рутинный анализ мочи может обнаружить скрытую гематурию или протеинурию, но чаще ничего не показывает. При повторяющихся инфекциях мочевых путей у пациентов с микротией исследование функции почек следует начать с УЗИ почек, прежде чем применять более инвазивные методики для выявления патологии. Из-за повышенной заболеваемости органов мочеполовой системы у больных с микротией целесообразно проводить периодическое обследование пациентов с помощью ультразвука.
Шейный отдел (шея) позвоночника
Если есть какие-либо подозрения на наличие неврологических нарушений необходимо пройти КТ, МРТ и соответствующее неврологическое обследование.
Другие патологии: расщелина губы и неба, сердце
У небольшого процента пациентов встречаются расщелины губы и неба и пороки сердечно-сосудистой системы. Последнее включает в себя дефект межпредсердной и межжелудочковой перегородок сердца, декстрокардию, транспозицию магистральных сосудов, трехкамерное сердце и незаращенный протока. Если отмечены какие-либо признаки или симптомы этих проблем с сердцем, педиатр должен проконсультироваться с кардиологом для соответствующего ведения пациентов.
Варианты микротии
Микротия варьируется от полного отсутствия ушных тканей (anotia) до практически нормального, но маленького, уха с суженным каналом. Между этими крайностями, каждый находит бесконечное разнообразие видов, наиболее распространенным из которых является вертикально-ориентированный бобовидной формы комочек. По данным статистики, микротия почти в два раза чаще встречается у мужчин, чем у женщин, соотношение правосторонней – левосторонней и двусторонней микротии составляет примерно 6: 3: 1.
В большинстве случаев, мочка дефектного уха смещается выше уровня противоположной нормальной стороны, но при этом при развитии происходит миграция пораженного уха в более низкое положение. Примерно одна треть пациентов имеют признаки гемифациальный микросомии, но учеными при обследовании пациентов с помощью рентгеновских исследований было показано, что недоразвитие костной ткани встречается во всех случаях.
Различают 3 степени микротии (I ст. – уменьшенная ушная раковина, II ст. – недоразвитие некоторых структур ушной раковины, III ст. – ушная раковина представлена в виде бобовидного комочка) и полное отсутствие ушной раковины - анотия.
Психическое здоровье и ушная раковина
По моему опыту, ребенок узнает, что он / она отличается от других примерно в возрасте до 3-3,5 лет. В классической ситуации родители находят своего ребенка у зеркала, сравнивающего разные стороны лица. Они начинают называть дефектное ухо, как "маленькое ухо" или "закрытое ухо". Лучше всего согласиться с ребенком, что он родился с одним большим ухом и одним маленьким, а когда он подрастет, можно будет маленькое ушко сделать больше, соответствующим другому. Затем дети должны воспитываться абсолютно нормально, не делая акцента на деформацию. Дети выглядят обеспокоенными по поводу микротии до 6-7 лет только в том случае, если родители передают свои опасения ребенку.
Исходя из моего опыта, нелеченный человек никогда не теряет желания стать полноценным и иметь исправленное ухо; на сегодняшний день самый взрослый пациент, которому я оперировала ушную раковину, 54 года.
В дополнение к таким функциям, как поддерживание очков, направление звуковой волны к барабанной перепонке, чтобы улучшить слух, ушные раковины позволяют нам выглядеть лучше и чувствовать себя как целостная личность. Это и является движущей причиной для хирургического создания наружного уха. Это психоэмоциональное эстетическое стремление восстановить самооценку путем восстановления симметричного, нормального образа. Совсем не являясь "косметической хирургией", исправление врожденной деформации позволяет человеку иметь нормальный образ самого себя, нормальную жизнь, и быть нормальным, продуктивным членом общества.
Если же наружное ухо не исправляется или достигается плохой результат вследствие отсутствия достаточного опыта у хирурга, состояние пациентов часто усугубляется, и страдание от низкой самооценки может длиться всю жизнь.
Из-за этих последствий, очень важно, чтобы педиатры направляли семью к опытному хирургу, даже если необходима далекая поездка для получения специализированной оценки.
Возраст для начала хирургического лечения.
Возраст, при котором следует начинать формирование ушной раковины, определяется как психологическими, так и физиологическими факторами. Восприятие изображения тела, как правило, начинает формироваться в возрасте около четырех-пяти лет, поэтому, в идеале, начать хирургическое лечение до поступления ребенка в школу, прежде чем он/она будут психологически травмированы от жестоких насмешек сверстников.
При реконструкции ушной раковины при помощи силиконового имплантата хирургическое вмешательство можно проводить с 6-7 лет. При использовании реберного аутохряща операция должна быть отложена до тех пор, пока размер грудной клетки, и в частности хрящевой части ребер, будет достаточных размеров, чтобы обеспечить изготовление каркаса ушной раковины, что часто достигается в возрасте около 9-10 лет.
В исследованиях определено, что ушные раковины активно продолжают свой рост в среднем до 10 лет, это примерно на 85%, поэтому целесообразно проводить оперативное лечение в этот период, чтобы сформированное наружное ухо достаточно долго сохраняло симметрию с противоположным.
При этом отмечено, что реберный хрящ имеет большой потенциал к росту и сохраняет его при операциях формирования наружного уха. Поэтому пластически восстановленные из реберного хряща ушные раковины растут вместе с окружающими мягкими тканями в течение жизни пациента.
Эталон новых стандартов! Беспрецедентная четкость, разрешение, сверхбыстрая обработка данных, а также исчерпывающий набор современных ультразвуковых технологий для решения самых сложных задач диагностики.
Синдром Тричера Коллинза (СТК) – это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза–Франческетти, синдром Франческетти–Цвалена–Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер – имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза–Франческетти [4, 5].
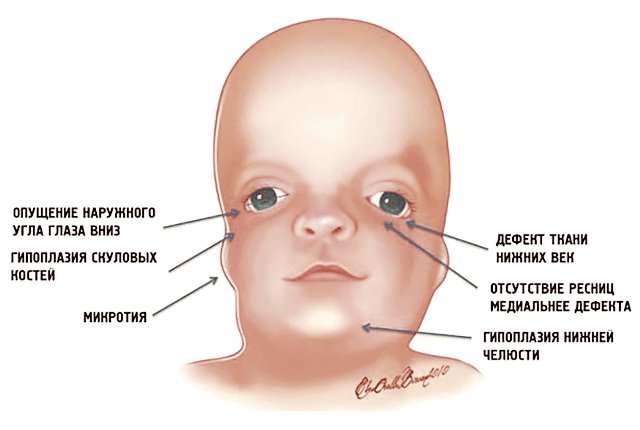
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса [1–6], также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости [1–8]. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев – это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 – q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4–6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип – мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности – редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким – 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12–13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2–4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).
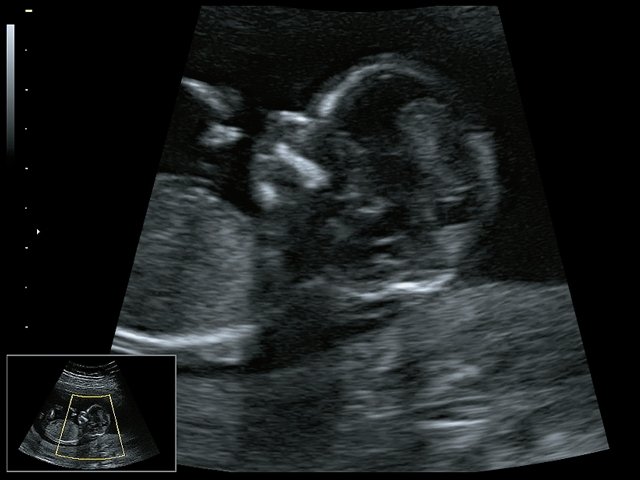
Рис. 2. Микрогнатия - сагиттальный скан в 2D, беременность 13 нед.
Наследственные факторы
По данным многих исследований существуют структурные, анатомические и генетические взаимосвязи между такими патологическими состояниями, как микротия, суженные и оттопыренные уши. Показано, что эти деформации взаимосвязаны и могут быть наследственными.
Выявлены как доминантные, так и рецессивные факторы наследования при глухоте, связанной с патологией ушных раковин. Деформации ушных раковин часто встречаются в семьях с синдромом Тричер-Коллинза (мандибулофациальный синостоз).
В исследовании семей с микротией у одного или обоих родителей с исключением хромосомных аберраций пришли к выводу, что наследование должно быть мультифакторным и что риск рецидива составляет в среднем 5,7% (от 3 до 8%) [95]. Среди ближайших родственников помимо различных дефектов и деформаций ушной раковины наблюдалось нарушение развития челюстей и лицевого нерва.
Специфические факторы
Среди ученых есть предположение, что ишемия тканей (снижение кровотока) вследствие закупорки артерии может стать причиной патологического развития ушных раковин. Это говорит о том, что причиной развития деформации чаще являются проблемы, возникающие во время развития плода, а не наследственные факторы. В поддержку этой теории выступает факт частого наличия микротии только у одного из близнецов. Детские дизморфологи заявляют, что многие виды врожденных уродств чаще встречаются при многоплодной беременности, и что это может быть вследствие явления под названием синдром "фетоплацентарного обкрадывания", когда плацента нормального двойника часто больше, чем плацента аномального, и, возможно, происходит усугубление кровообращения одного из близнецов, развитие которого затем нарушается.
Хорошо известно, что появление глухоты и иногда микротии происходит в результате краснухи в течение первого триместра беременности. Кроме того, использование некоторых лекарств в этот критический период тоже может стать причиной врожденных заболеваний ( например, талидомида, роаккутан (Accutane), кломид, ретиноевая кислота).
Влияние различных внешних факторов во время первого триместра беременности на развитие микротии у ребенка не однозначно, в 1-4% случаев это могут быть простудные заболевания, корь, травмы, облучение, месячные во время первого триместра, гиперемезис (чрезмерная рвота), наличие диабета, эмоциональные перегрузки, употребление алкоголя, лекарства от тошноты.
В заключение, развитие микротии – это, как правило, случайное спорадическое событие, и важно, чтобы родители понимали, что деформация не была вызвана какими-либо действиями матери до или во время беременности, и что риск рецидива в семье составляет около пяти процентов, или, иначе говоря, один из двадцати.
Ассоциированные патологии
Эмбриологическое развитие подразумевает, что микротия обычно сопровождается патологией среднего уха. При классической микротии обычно присутствует атрезия слухового канала и нарушения слуховых косточек - Синдром Конигсмарка (микротия, атрезия наружного слухового прохода и кондуктивная тугоухость).
Деформация среднего уха может варьироваться от суженного канала и незначительных нарушений слуховых косточек до расплавленных гипоплазированных слуховых косточек и недостаточной аэрации сосцевидной кости. Следует обратить внимание, что, поскольку у пациентов с атрезией есть слуховые трубы, как и у всех, они могут спровоцировать воспаление среднего уха (средний отит) даже если они не имеют наружный слуховой проход. Поэтому, если этот диагноз невозможно подтвердить с помощью отоскопии, при наличии подозрения на средний отит в деформированном ухе, разумно назначить антибиотики.
Патология челюстно-лицевой области
Так как ушная раковина развивается из тканей жаберных дуг, не удивительно, что у значительного процента пациентов с микротией обнаруживают дефицитные компоненты лица, которые происходят из этих эмбриональных элементов. Проявление в виде уменьшенной половины лица, состояние, известное как гемифациальная микросомия – это, в основе, недоразвитие челюстей и вышележащих мягких тканей. Наиболее полное генетическое проявление этого состояния включает в себя дефекты наружного и среднего уха, гипоплазию верхней и нижней челюстей, скуловой и височной костей, макростому и боковые щели лица, парез лицевого нерва, атрофии мышц лица и околоушной железы, даже небные мышцы могут быть ослаблены на вовлеченной стороне.
Почки и мочевыводящие пути
Нарушения урогенитального тракта увеличиваются при наличии деформаций ушной раковины, особенно когда пациент страдает от других проявлений недоразвития лица. У некоторых пациентов может наблюдаться недоразвитие половых органов (гипоспадия, агенезия женских половых органов), различные нарушения почек (подковообразная почка, односторонняя почечная агенезия, тазовое расположение почки и т.п.). Однако эти нарушения не вызывают жизненно-опасных нарушений работы органов мочеполовой системы. Рутинный анализ мочи может обнаружить скрытую гематурию или протеинурию, но чаще ничего не показывает. При повторяющихся инфекциях мочевых путей у пациентов с микротией исследование функции почек следует начать с УЗИ почек, прежде чем применять более инвазивные методики для выявления патологии. Из-за повышенной заболеваемости органов мочеполовой системы у больных с микротией целесообразно проводить периодическое обследование пациентов с помощью ультразвука.
Шейный отдел (шея) позвоночника
Если есть какие-либо подозрения на наличие неврологических нарушений необходимо пройти КТ, МРТ и соответствующее неврологическое обследование.
Другие патологии: расщелина губы и неба, сердце
У небольшого процента пациентов встречаются расщелины губы и неба и пороки сердечно-сосудистой системы. Последнее включает в себя дефект межпредсердной и межжелудочковой перегородок сердца, декстрокардию, транспозицию магистральных сосудов, трехкамерное сердце и незаращенный протока. Если отмечены какие-либо признаки или симптомы этих проблем с сердцем, педиатр должен проконсультироваться с кардиологом для соответствующего ведения пациентов.
Варианты микротии
Микротия варьируется от полного отсутствия ушных тканей (anotia) до практически нормального, но маленького, уха с суженным каналом. Между этими крайностями, каждый находит бесконечное разнообразие видов, наиболее распространенным из которых является вертикально-ориентированный бобовидной формы комочек. По данным статистики, микротия почти в два раза чаще встречается у мужчин, чем у женщин, соотношение правосторонней – левосторонней и двусторонней микротии составляет примерно 6: 3: 1.
В большинстве случаев, мочка дефектного уха смещается выше уровня противоположной нормальной стороны, но при этом при развитии происходит миграция пораженного уха в более низкое положение. Примерно одна треть пациентов имеют признаки гемифациальный микросомии, но учеными при обследовании пациентов с помощью рентгеновских исследований было показано, что недоразвитие костной ткани встречается во всех случаях.
Различают 3 степени микротии (I ст. – уменьшенная ушная раковина, II ст. – недоразвитие некоторых структур ушной раковины, III ст. – ушная раковина представлена в виде бобовидного комочка) и полное отсутствие ушной раковины - анотия.
Психическое здоровье и ушная раковина
По моему опыту, ребенок узнает, что он / она отличается от других примерно в возрасте до 3-3,5 лет. В классической ситуации родители находят своего ребенка у зеркала, сравнивающего разные стороны лица. Они начинают называть дефектное ухо, как "маленькое ухо" или "закрытое ухо". Лучше всего согласиться с ребенком, что он родился с одним большим ухом и одним маленьким, а когда он подрастет, можно будет маленькое ушко сделать больше, соответствующим другому. Затем дети должны воспитываться абсолютно нормально, не делая акцента на деформацию. Дети выглядят обеспокоенными по поводу микротии до 6-7 лет только в том случае, если родители передают свои опасения ребенку.
Исходя из моего опыта, нелеченный человек никогда не теряет желания стать полноценным и иметь исправленное ухо; на сегодняшний день самый взрослый пациент, которому я оперировала ушную раковину, 54 года.
В дополнение к таким функциям, как поддерживание очков, направление звуковой волны к барабанной перепонке, чтобы улучшить слух, ушные раковины позволяют нам выглядеть лучше и чувствовать себя как целостная личность. Это и является движущей причиной для хирургического создания наружного уха. Это психоэмоциональное эстетическое стремление восстановить самооценку путем восстановления симметричного, нормального образа. Совсем не являясь "косметической хирургией", исправление врожденной деформации позволяет человеку иметь нормальный образ самого себя, нормальную жизнь, и быть нормальным, продуктивным членом общества.
Если же наружное ухо не исправляется или достигается плохой результат вследствие отсутствия достаточного опыта у хирурга, состояние пациентов часто усугубляется, и страдание от низкой самооценки может длиться всю жизнь.
Из-за этих последствий, очень важно, чтобы педиатры направляли семью к опытному хирургу, даже если необходима далекая поездка для получения специализированной оценки.
Возраст для начала хирургического лечения.
Возраст, при котором следует начинать формирование ушной раковины, определяется как психологическими, так и физиологическими факторами. Восприятие изображения тела, как правило, начинает формироваться в возрасте около четырех-пяти лет, поэтому, в идеале, начать хирургическое лечение до поступления ребенка в школу, прежде чем он/она будут психологически травмированы от жестоких насмешек сверстников.
При реконструкции ушной раковины при помощи силиконового имплантата хирургическое вмешательство можно проводить с 6-7 лет. При использовании реберного аутохряща операция должна быть отложена до тех пор, пока размер грудной клетки, и в частности хрящевой части ребер, будет достаточных размеров, чтобы обеспечить изготовление каркаса ушной раковины, что часто достигается в возрасте около 9-10 лет.
В исследованиях определено, что ушные раковины активно продолжают свой рост в среднем до 10 лет, это примерно на 85%, поэтому целесообразно проводить оперативное лечение в этот период, чтобы сформированное наружное ухо достаточно долго сохраняло симметрию с противоположным.
При этом отмечено, что реберный хрящ имеет большой потенциал к росту и сохраняет его при операциях формирования наружного уха. Поэтому пластически восстановленные из реберного хряща ушные раковины растут вместе с окружающими мягкими тканями в течение жизни пациента.

Синдром Гольденхара
Описание
Синдром Гольденхара. Это редкое врожденное заболевание, которое проявляется множественными пороками развития, выраженным клиническим полиморфизмом. Возникает в результате мутации генов, расположенных в 5, 14, 20 хромосомах. Для синдрома характерны различные аномалии лицевого скелета, патологии органов чувств, часто заболевание сопровождается умственной отсталостью. В план обследования входят генетические тесты для верификации диагноза, лабораторные и инструментальные методы с учетом основных клинических признаков. Лечение поддерживающее, многим пациентам требуются слуховые аппараты, комплексная нейрореабилитация.
Дополнительные факты

Синдром Гольденхара
Причины
Заболевание возникает при дифференцировке 1-2-й. Нарушение жаберной дуги вызвано мутациями генов в 14-й (Локус 14q32), 5-й (Локус 5p15), 20-й хромосоме (ген MYT1, Локус q13,33). В 98% случаев синдрома роль наследственности не прослеживается. Однако около 2% пациентов имеют родственников с аналогичными клиническими симптомами. В литературе описаны случаи аутосомно-доминантного и аутосомно-рецессивного наследования.
Основными предрасполагающими факторами являются:
• воздействие тератогенных факторов на ранних сроках беременности.
• мать больна сахарным диабетом, имеет избыточный вес.
• Предыдущие искусственные аборты и выкидыши.
Патогенез
Механизмы формирования фенотипических изменений, характерных для синдрома Гольденхара, продолжают изучаться. Сегодня основной теорией является теория пороков развития структур лица на ранних сроках беременности (3-8 неделя эмбриогенеза). Предположительно, в этот период в результате совокупного воздействия неблагоприятных внешних факторов и генетических аномалий нарушается дифференциация первой-второй парных жаберных дуг.
В результате лобные, нижнечелюстные и верхнечелюстные эктодермальные отростки, отходящие от первой жаберной дуги, и ушные раковины, образованные 1-й и 2-й жаберными дугами, развиваются асимметрично. После рождения ребенка это проявляется специфическими изменениями верхней и нижней челюсти, глаз и глазниц, мимических и жевательных мышц, структур наружного и среднего уха, неправильным прикусом, дефицитом мягких тканей.
Клиническая картина
Основные признаки синдрома Гольденхара - аномалии строения лица, которое в 70% случаев имеет правосторонний характер. У всех пациентов наблюдается асимметрия, недоразвитие нижней челюсти, уменьшение размеров, деформация или отсутствие ушных раковин. Патологии органа слуха часто бывают односторонними, сопровождаются атрезией слухового прохода, преаурикулярными разрастаниями. Иногда образуется добавочная рудиментарная ушная раковина.
Характерно также уменьшение размеров глазных яблок (микрофтальм), косоглазие, атрезия радужной оболочки, катаракта. Около 50% пациентов с болезнью Гольденхара имеют высокое готическое небо, широкий рот (макростомия), поврежденный язык, ненормальный прикус и отсутствующую часть зубов. Косолапость, аномалии позвоночника (сколиоз, расщелина позвоночника), искривление ребер отмечаются в 40% случаев. У 30% пациентов развиваются врожденные патологии внутренних органов: пороки сердца, гипоплазия легких, дисплазия почек.
Возможные осложнения
Наиболее опасным последствием синдрома Гольденхара является умственная отсталость, вызванная потерей слуха и снижением остроты зрения. При этом у большинства детей наблюдается нормальное функционирование нервной системы, но сенсорная недостаточность приводит к затруднениям в речевом и психомоторном развитии. Осложненное течение синдрома протекает при тяжелых врожденных пороках сердца, легких и почек.
Диагностика
В большинстве случаев предварительный диагноз ставится на основании характерных фенотипических признаков: деформации уха, асимметрии лица, патологии развития нижней челюсти. Для подтверждения синдрома Гольденхара должна быть назначена консультация генетика, проводится комплекс диагностических мероприятий:
• Генетический анализ. Поиск конкретной генетической мутации 14q32, 5p15, MYT1 позволяет на 100% подтвердить диагноз. Исследование проводится в специальных генетических лабораториях с использованием методов секвенирования экзонов и флуоресцентной гибридизации.
• Аудиометрия. Динамическая оценка слуха необходима всем пациентам для своевременного выявления кондуктивной тугоухости и принятия мер по ее коррекции. Тщательное обследование у отоларинголога может включать измерение импеданса, оценку мышечного рефлекса стремени.
• Неврологическое обследование. Консультация детского невролога необходима для определения причин задержки психомоторного развития, оценки работы центральной и периферической нервной системы. При наличии доказательств пациента направляют на повторное обследование к психиатру.
• Инструментальный дисплей. Для подтверждения или исключения соматических дефектов при синдроме Гольденхара проводят рентген грудной клетки, эхокардиографию и УЗИ органов брюшной полости. Для исследования центральной нервной системы используется КТ или МРТ головного мозга.
Лечение
Поскольку потеря слуха является основной проблемой, слуховые аппараты рекомендуются с раннего возраста для предотвращения глухоты и связанной с ней умственной отсталости. Кроме того, пациент осматривается пластическим хирургом с целью коррекции типичных внешних проявлений синдрома, по возможности скорректировать асимметрию и гармонизировать черты лица.
Пациенты, у которых диагностирована болезнь Гольденхара, подлежат пожизненному наблюдению. Они проходят регулярные осмотры у офтальмолога, отоларинголога, невролога и других специалистов, чтобы следить за своим здоровьем. Курсы поддерживающей терапии включают несколько направлений лечения, основными из которых являются:
• Метаболическая терапия. Для правильного развития ЦНС, улучшения познавательных способностей используются ноотропы, витамины группы В.
• Нейрореабилитация. Для улучшения речевого развития рекомендуются длительные курсы с логопедами. По показаниям проводится психологическая коррекция.
• Специальные занятия. Пациентам с признаками тяжелой умственной отсталости необходимо продолжить обучение в коррекционных классах у дефектологов, глухих педагогов.
Прогноз
При отсутствии врожденных соматических пороков развития у пациента и достаточном уровне интеллекта прогноз благоприятный, большинство пациентов доживают до зрелого и старческого возраста. Случаи синдрома Гольденхара, которые сопровождаются кардиологическими, легочными или нефрологическими отклонениями, вызывают беспокойство, также устанавливается неблагоприятный прогноз при тяжелой умственной отсталости.
Основа профилактики синдрома - внутриутробная защита плода. Критическим периодом является первый триместр, так как именно в этот период возникают характерные костные аномалии. Беременным рекомендуется избегать контакта с химическими веществами, рентгеновскими лучами, а также соблюдать противоэпидемические меры для защиты от вирусных инфекций.
Список литературы
1. Клинический случай синдрома Гольденхара в психиатрической практике/ А.В. Ковалева// Acta Biomedica Scientifica. — 2020. — №3.
2. Клинический случай синдрома Гольденхара у новорожденного/ Л.Г. Киселева, Л.П. Мокеева, Ю.С. Тишкова, Н.В. Павловский// Вятский медицинский вестник. — 2015. — №46.
3. Диагностика синдрома Гольденхара в периоде новорожденности/ С.И. Мизинцева// Бюллетень Северного государственного медицинского университета. — 2012.
4. Особенности общеклинических проявлений синдрома Гольденхара/ И.А. Карякина// Системная интеграция в здравоохранении. — 2010. — №2.
Читайте также: