
Эпидермолиз передается ли по наследству
Обновлено: 02.07.2024
Механобуллезные заболевания (буллезный эпидермолиз, БЭ) представляют собой гетерогенную группу пузырных дерматозов, для которых характерно образование очагов после травмы кожи. Варианты буллезного эпидермолиза (БЭ) дифференцируют по типу наследования, мутациям специфических генов, клинической картине, гистопатологическим и биологическим маркерам. Хотя пузыри могут появиться при рождении или в неонатальный период, первая манифестация легких форм с локализованными очагами часто запаздывает до наступления старшего детского или взрослого возраста.
Классификация буллезного эпидермолиза (БЭ) на три основных типа учитывает слой, в котором происходит образование пузыря. При простом буллезном эпидермолизе (БЭ) пузыри образуются внутри эпидермиса, и клинически заболевание протекает в легкой форме. При пограничном БЭ пузыри образуются в зоне дермо-эпидермального соединения, обычно на уровне светлой пластинки базальной мембраны. Хотя заболевание у пациентов может протекать в легкой форме, сравнимой с простым БЭ, у некоторых детей образование пузырей прогрессирует с поражением слизистых оболочек и желудочно-кишечного тракта (ЖКТ), ребенок утрачивает жизненную силу, становится пассивным, развивается сепсис с летальным исходом в первые 2 года жизни.
Для дистрофического буллезного эпидермолиза (БЭ) характерны рубцующиеся пузыри, которые образуются в дерме, под зоной базальной мембраны. Пузыри могут располагаться на ладонях и подошвах или распространяются с поражением зубо-челюстной системы, ногтей, дыхательных путей и пищевода.
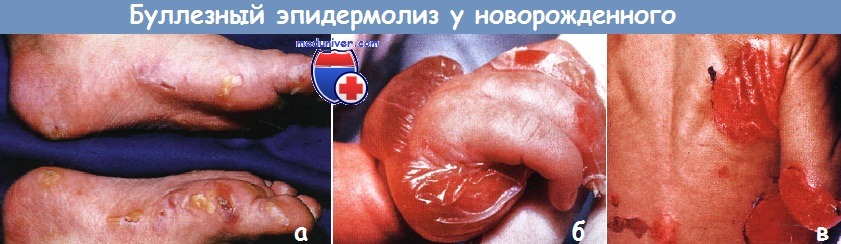
а - простой буллезный эпидермолиз (БЭ). Многочисленные пузыри образуются, прежде всего, на участках давления в области кистей и стоп.
б, в - пограничный буллезный эпидермолиз (БЭ). У этого ребенка обширное поражение наблюдалось при рождении.
(б) Обратите внимание на эрозии и крупный интактный пузырь над большим пальцем и тыльной стороной кисти.
(в) Крупные оголенные участки на спине и ягодицах.
После исключения инфекции у детей с рецидивирующими пузырями следует подозревать буллезный эпидермолиз (БЭ). Семейный анамнез и обследование других членов семьи помогают установить клинический диагноз. Биопсия кожи для иммунологического картирования и/или электронной микроскопии обеспечивает точную идентификацию плоскостного слоя кожи, в котором образуется пузырь. С семьей следует обсудить необходимость генетического консультирования и пренатальной диагностики, особенно в случае тяжелых вариантов заболевания. При некоторых вариантах возможен прена-тальный диагноз с использованием генных маркеров на амниоцитах или с применением электронной микроскопии биопсий кожи плода.
Для детей без буллезного эпидермолиза (БЭ) в семейном анамнезе сделать прогноз в период новорожденности трудно. Младенцы с дистрофическим буллезным эпидермолизом (БЭ) могут развиваться очень хорошо, в то время как у детей с пограничным вариантом заболевание после периода стабилизации может прогрессивно ухудшаться. В такой ситуации врачу следует успокоить родителей и отложить обсуждение прогноза на время после периода наблюдения.
Терапия зависит от тяжести буллезного эпидермолиза (БЭ). При легких вариантах пациенты учатся избегать травм, которые способствуют образованию пузырей. При боли и крупных пузырях можно аккуратно вскрыть покрышку очага или вырезать в ней квадратное окошко и наложить местную мазь с антибиотиком и стерильный бинт. Пластыри приклеивают с повязки на повязку, избегая контакта с кожей.
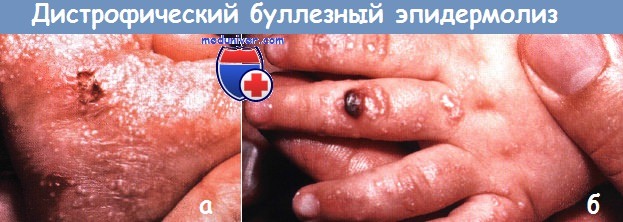
Дистрофический буллезный эпидермолиз (БЭ):
(а) Пузыри, эрозии и сотни милиумов наблюдаются на ступне и лодыжке и на
(б) кисти у этого новорожденного
При тяжелом течении врач должен организовать мультидисциплинарный подход к ведению пациентов. В лечении ребенка должны принимать участие дерматолог, офтальмолог, гастроэнтеролог, отоларинголог, пластический хирург, торакальный хирург, стоматолог и физиотерапевт.
Профилактика включает избегание травм кожи и слизистых оболочек и раннюю терапию инфекций топическими или системными антибиотиками. Может потребоваться восполнение железа вследствие хронических потерь крови через кожу. Для сохранения влаги, уменьшения боли и ускоренного заживления эрозий и язв применяются повязки Adaptic®, Telfa® и бинт Vaseline®. Местные повязки аккуратно фиксируют, обертывая чистым марлевым или самофиксирующимся бинтом Kling®, и удаляют с помощью замачивания, не разрывая свежую грануляционную ткань.
Полупроницаемые повязки (N-ter-Face®, Ensure®, Vigilon®, Mepilex, Mepitel) и герметичные повязки (Duoderm®, Comfeel®) помогают в терапии стойких ран. Хорошее питание обязательно для заживления кожи и нормального роста и развития.
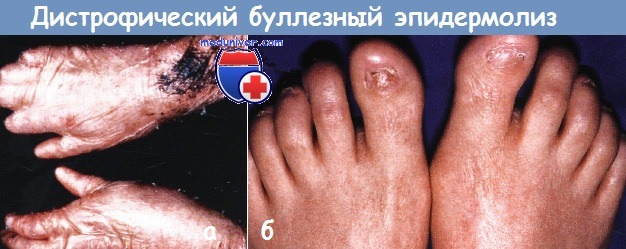
Дистрофический буллезный эпидермолиз (БЭ):
а - у этого ребенка тяжелое рубцевание сковало пальцы и привело к синдактилии
б - рецидивирующие пузыри привели к рубцеванию и перманентной утрате ногтей у этого подростка.
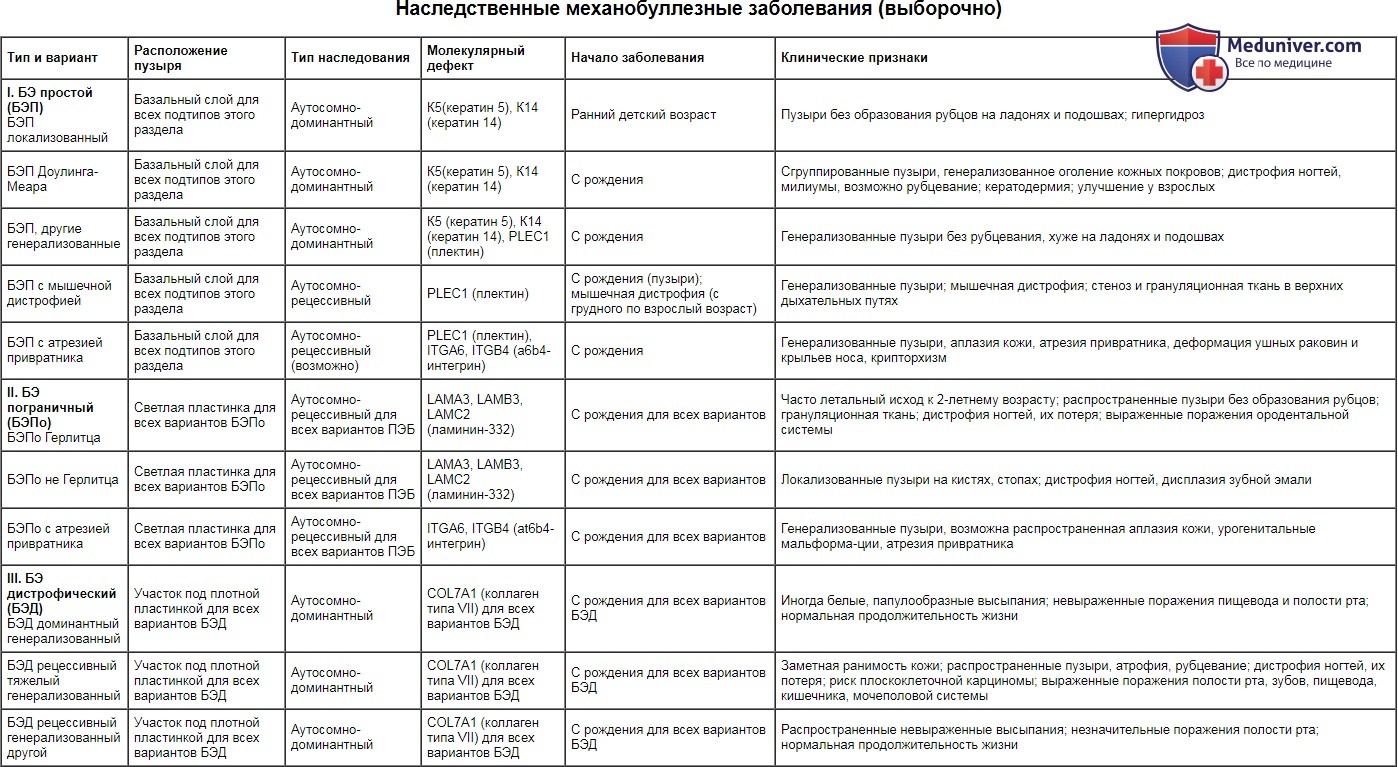
Наследственные механобуллезные заболевания (выборочно)
| Тип и вариант | Расположение пузыря | Тип наследования | Молекулярный дефект | Начало заболевания | Клинические признаки |
| I. БЭ простой (БЭП) БЭП локализованный | Базальный слой для всех подтипов этого раздела | Аутосомно-доминантный | К5(кератин 5), К14 (кератин 14) | Ранний детский возраст | Пузыри без образования рубцов на ладонях и подошвах; гипергидроз |
| БЭП Доулинга-Меара | Базальный слой для всех подтипов этого раздела | Аутосомно-доминантный | К5(кератин 5), К14 (кератин 14) | С рождения | Сгруппированные пузыри, генерализованное оголение кожных покровов; дистрофия ногтей, милиумы, возможно рубцевание; кератодермия; улучшение у взрослых |
| БЭП, другие генерализованные | Базальный слой для всех подтипов этого раздела | Аутосомно-доминантный | К5 (кератин 5), К14 (кератин 14), PLEC1 (плектин) | С рождения | Генерализованные пузыри без рубцевания, хуже на ладонях и подошвах |
| БЭП с мышечной дистрофией | Базальный слой для всех подтипов этого раздела | Аутосомно-рецессивный | PLEC1 (плектин) | С рождения (пузыри); мышечная дистрофия (с грудного по взрослый возраст) | Генерализованные пузыри; мышечная дистрофия; стеноз и грануляционная ткань в верхних дыхательных путях |
| БЭП с атрезией привратника | Базальный слой для всех подтипов этого раздела | Аутосомно-рецессивный (возможно) | PLEC1 (плектин), ITGA6, ITGB4 (а6b4-интегрин) | С рождения | Генерализованные пузыри, аплазия кожи, атрезия привратника, деформация ушных раковин и крыльев носа, крипторхизм |
| II. БЭ пограничный (БЭПо) БЭПо Герлитца | Светлая пластинка для всех вариантов БЭПо | Аутосомно-рецессивный для всех вариантов ПЭБ | LAMA3, LAMB3, LAMC2 (ламинин-332) | С рождения для всех вариантов | Часто летальный исход к 2-летнему возрасту; распространенные пузыри без образования рубцов; грануляционная ткань; дистрофия ногтей, их потеря; выраженные поражения ородентальной системы |
| БЭПо не Герлитца | Светлая пластинка для всех вариантов БЭПо | Аутосомно-рецессивный для всех вариантов ПЭБ | LAMA3, LAMB3, LAMC2 (ламинин-332) | С рождения для всех вариантов | Локализованные пузыри на кистях, стопах; дистрофия ногтей, дисплазия зубной эмали |
| БЭПо с атрезией привратника | Светлая пластинка для всех вариантов БЭПо | Аутосомно-рецессивный для всех вариантов ПЭБ | ITGA6, ITGB4 (аt6b4-интегрин) | С рождения для всех вариантов | Генерализованные пузыри, возможна распространенная аплазия кожи, урогенитальные мальформа-ции, атрезия привратника |
| III. БЭ дистрофический (БЭД) БЭД доминантный генерализованный | Участок под плотной пластинкой для всех вариантов БЭД | Аутосомно-доминантный | COL7A1 (коллаген типа VII) для всех вариантов БЭД | С рождения для всех вариантов БЭД | Иногда белые, папулообразные высыпания; невыраженные поражения пищевода и полости рта; нормальная продолжительность жизни |
| БЭД рецессивный тяжелый генерализованный | Участок под плотной пластинкой для всех вариантов БЭД | Аутосомно-доминантный | COL7A1 (коллаген типа VII) для всех вариантов БЭД | С рождения для всех вариантов БЭД | Заметная ранимость кожи; распространенные пузыри, атрофия, рубцевание; дистрофия ногтей, их потеря; риск плоскоклеточной карциномы; выраженные поражения полости рта, зубов, пищевода, кишечника, мочеполовой системы |
| БЭД рецессивный генерализованный другой | Участок под плотной пластинкой для всех вариантов БЭД | Аутосомно-доминантный | COL7A1 (коллаген типа VII) для всех вариантов БЭД | С рождения для всех вариантов БЭД | Распространенные невыраженные высыпания; незначительные поражения полости рта; нормальная продолжительность жизни |
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Врожденный буллезный эпидермолизпредставляет собой гетерогенную группу заболеваний, которые развиваются вследствие врожденного дефекта в генах, кодирующих разные протеины дермо-эпидермального соединения, и проявляются неустойчивостью кожного покрова к механическим воздействиям с развитием травмо-индуцированных пузырей и эрозий.Спектр клинических проявлений велик: от локализованных пузырей на коже конечностей до генерализованных пузырей на коже и слизистой оболочке полости рта с вовлечением в патологический процесс других органов и тканей.
В большинстве случаев диагноз врожденного буллезного эпидермолиза при наличии характерной клиники не вызывает сомнения. Однако при отсутствии семейной истории и/или наличии атипичных клинических проявлений требует от врача клинический опыт и знаний. Так, в неонатальный период все типы врожденного буллезного эпидермолиза имеют сходную клиническую картину и не следует пытаться новорожденному определить тип заболевания. Только в течение жизни в определенные промежутки времени проявляются дополнительные клинические признаки, характерные для каждого типа и подтипа врожденного буллезного эпидермолиза.
Для подтверждения клинического диагноза, определения типа и подтипа врожденного буллезного эпидермолизаприменяют гистологический, электронно-микроскопический и иммуногистохимический методы исследования.
По уровню отслойки эпидермиса и формированию пузырей различают 4 основных типа врожденного буллезного эпидермолиза: простой, пограничный, дистрофический и Киндлер-синдром. Для простого типа врожденного буллезного эпидермолизахарактерны внутриэпидермальные пузыри на уровне базального и супрабазальных слоев эпидермиса. При пограничном и дистрофическом типах врожденного буллезного эпидермолизапузыри возникают на уровне базальной мембраны эпидермиса с вовлечением в патологический процесс laminalucida и sublaminadensa соответственно. При Киндлер-синдроме наблюдают множественную отслойку эпидермиса и дермына разных уровнях. При этом оценку состояния специфических структур ткани (филаменты кератина; полудесмосомы; якорные фибриллы; sublaminadensa) проводят с помощью трансмиссионной электронной микроскопии.
В настоящее время наиболее ценным методом диагностики врожденного буллезного эпидермолиза является иммуногистохимический метод с использованием моноклональных антител, направленных к отдельным детерминантам соединений организма, которые в норме экспрессируютсяв коже. При вовлечении в патологический процесс специфических белков (антигенов) типичная иммуногистохимическая реакция отсутствует. На основании идентификации отсутствующего белкаустанавливается тип врожденного буллезного эпидермолиза. При простом типеврожденного буллезного эпидермолизаантигенами-мишенями являются десмоплакин, плакофиллин-1, цитокератины 5 и 14, плектин, α6β4интегрин; при пограничном типе - ламинин-332, коллаген XVII типа, α6β4-интегрин; при дистрофическом типе - коллаген VII типа; при Киндлер-синдроме - киндлин-1.
По совокупности полученных данных (анамнез, клинико-морфологическая картина, лабораторные методы исследования) диагностируют тип и подтип врожденного буллезного эпидермолиза.
Основными принципами лечения врожденного буллезного эпидермолизаявляются замещение потери тканевой жидкости и использование препаратов, обладающих ранозаживляющим эффектом. Вопрос применения препаратов глюкокортикостероидного ряда дискутабелен. Тем не менее, наблюдаемые клинические случаи и результаты иммуногистохимического исследования диктуют целесообразность включения их в терапию при данной патологии. Фиксация иммунных комплексов (IgG, C3 компонент комплемента) и фибрин в зоне базальной мембраны эпидермиса и волосяных фолликулов свидетельствует о вовлечении в патологический процесс изначально дефектной базальной мембраны как мишени для антител. В результате развивается вторичный аутоиммунный процесс.
Специфической терапии врожденного буллезного эпидермолизав настоящее время нет.Внедрение молекулярно-биологическихметодов исследования, позволившихвыявить причины возникновения каждого типа и подтипа врожденного буллезного эпидермолиза,открывает новые возможности в создании перспективных методов лечения.Цельюэтих методов являетсявосстановление синтеза белковых компонентов и функции структурных элементов дермо-эпидермального соединения.
В качестве терапевтического лечения рассматриваютсяген-замещение, трансплантация аллогенных фибробластов (для обеспечения нормального синтеза коллагена VII типа), трансплантация клеток костного мозга и инфузия рекомбинантного протеина (коллаген VII типа). Генная и клеточная терапия, имея стратегическое значение, открывает новые перспективы в лечении врожденного буллезного эпидермолиза.
Учитывая клинические проявления и особенности течения болезни, особое внимание уделяют профилактическим мероприятиям по предотвращению возникновений механических повреждений покровного эпителия (эпидермис, слизистые оболочки) и вторичной инфекции. Высокий риск развития специфических экстракожных осложнений требует постоянного наблюдения и отсмежных специалистов для их выявления и назначения соответствующих терапевтических и/или хирургических вмешательств. Так, поражение роговицы глаза, приводящее к развитию рубцов и нарушению зрения, требует своевременного офтальмологического вмешательства. Наличие стриктур пищевода -баллонное его расширение (бужирование) и введения дополнительного парентерального питания. Деформация рук и стоп - хирургического вмешательства.Плоскоклеточный рак кожи - активное консервативное и/или хирургическое лечение с последующим мониторингом с целью исключения локального или регионального рецидива. В ряде случаев необходим постоянный лабораторный мониторинг для исключения остеопороза, остеопении, анемии, амилоидоза, дилатационнойкардиомиопатиии других патологий со стороны внутренних органов и систем.
Прогноз врожденного буллезного эпидермолизазначительно варьирует и зависит как от типа и подтипа заболевания, так и от общего состояния пациента.Пациенты, страдающие простыми доминантно-дистрофическим типами врожденного буллезного эпидермолиза, имеют, как правило,благоприятное течение болезни с сохранением социальной активности. В противоположность, пациенты, страдающие пограничным типом врожденного буллезного эпидермолизаимеют высокий риск возникновения смерти в течение первых пяти лет жизни, а пациенты с рецессивно-дистрофическим типом врожденного буллезного эпидермолиза - в молодом возрасте от метастазов плоскоклеточного рака кожи.
Таким образом, пациенты с врожденным буллезным эпидермолизом нуждаются в междисциплинарном подходе, в основе которого лежит защита чувствительной кожи от травмы с использованием разных перевязочных средств, медикаментозных и/или хирургических вмешательств с целью коррекции возможных осложнений.За последние два десятилетия достигнутые успехи в изучении патогенезаэтой тяжелой врожденной патологии на молекулярно-биологическом уровнепозволяют ученым возлагать большие надежды насозданиеновых методов, основанных на молекулярно-генной инженерии

Приобретенный буллезный эпидермолиз впервые описан во второй половине прошлого столетия и получил свое название в связи с клиническим сходством с дистрофической формой врожденного буллезного эпидермолиза. Последний развивается у детей вследствие врожденного дефекта в гене, кодирующем коллаген VII типа (якорные фибриллы), и проявляется неустойчивостью кожного покрова к механическим воздействиям с развитием травмо-индуцированных пузырей с образованием рубцов и милиум.
В основе диагностики приобретенного буллезного эпидермолиза лежит возникновение спонтанных или травмо-индуцированных пузырей в зрелом возрасте больного с отсутствием семейной предрасположенности и исключением других буллезных дерматозов. Различают два варианта приобретенного буллезного эпидермолиза: механобуллезный (классический или невоспалительный) и пемфигоидо-подобный (воспалительный).
При классическом варианте приобретенного буллезного эпидермолиза главным клиническим отличительным признаком является первичное возникновение пузырей и эрозивных дефектов на месте травм без воспалительного процесса или с минимальным его проявлением. Это связано с хрупкостью кожного покрова, особенно в выступающих местах и местах сдавления (локтевые и коленные сгибы, своды стоп). На месте эпителизации эрозивных дефектов формируются рубцовая атрофия, милиумы и/или эритема. Кроме того, у больных классическим приобретенным буллезным эпидермолизом патологический процесс первоначально может возникнуть на слизистой оболочке полости рта (слизистая щек, задняя стенка глотки, боковая поверхность языка, десна) и/или красной каймы нижней губы. Вовлечение в патологический процесс слизистых оболочек (полости рта, носа, пищевода, конъюнктивы, ануса, влагалища) может напоминать картину рубцующегося пемфигоида. Возможно развитие и стеноза пищевода как угрожающее жизни осложнение данного варианта приобретенного буллезного эпидермолиза. На рис. 15 представлены клинические признаки классического варианта приобретенного буллезного эпидермолиза.
При воспалительном варианте приобретенного буллезного эпидермолиза пузыри возникают на воспаленном участке кожи без травматизации (на открытых участках кожи тела). Кроме пузырей наблюдаются везикулы, склонные к слиянию с образованием гирляндоподобных элементов на эритематозном фоне, уртикарные и папулезные высыпания с вовлечением в патологический процесс слизистой оболочки полости рта. В ряде случаев поражается слизистая оболочка носа в виде геморрагических корочек и конъюнктивы (конъюнктивит). Картина может напоминать буллезный пемфигоид, реже герпетиформный дерматит Дюринга. На рис. 16 представлены клинические признаки воспалительного варианта приобретенного буллезного эпидермолиза.
В случаях приобретенного буллезного эпидермолиза паранеопластического генеза клиническая картина может быть представлена пузырями и обширными эрозивными дефектами на коже туловища и конечностей (рис. 17) с вовлечением в патологический процесс слизистой оболочки полости рта. Картина может напоминать вульгарную пузырчатку с положительным симптомом Никольского.
Одним из основных патогенетических факторов, способствующих развитию приобретенного буллезного эпидермолиза, признают наличие аутоантител к одному из компонентов базальной мембраны кожи человека и животных. Антитела не являются специфичными только для антигенов базальной мембраны эпидермиса, они также взаимодействуют с антигенами базальных мембран слизистой оболочки полости рта, пищевода, влагалища. Однако указанные антитела не реагируют с антигенами базальных мембран кровяносных сосудов, печени и эпителия паренхимы почки.
Локализация антигенов, к которым направлены антитела, совпадает с локализацией первичных деструктивных изменений. Образование подэпидермального пузыря (рис. 18, a) при приобретенном буллезном эпидермолизе начинается с расщепления дермо-эпидермального соединения под lamina densa или в верхних отделах дермы под комплексом lamina densa-якорные фибриллы (электронно-микроскопические данные). Установлено, что антигеном-мишенью для аутоантител является коллаген VII типа в пределах якорных фибрилл, которые проходят перпендикулярно от lamina densa к сосочковому слою дермы.
Ранее признанным аутоантигеном приобретенного буллезного эпидермолиза считалась α-цепь молекулярной массой 290 кД коллагена VII типа. Дальнейшее изучение строения коллагена данного типа на молекулярно-биологическом уровне позволило выявить, что антигеном-мишенью для этого буллезного дерматоза является ряд эпитопов молекул коллагена. А именно NC1 домен содержит главные иммунодоминирующие антигенные эпитопы для аутоантител приобретенного буллезного эпидермолиза. Этот домен может способствовать соединению коллагена VII типа с другими компонентами базальной мембраны и матрикса. Возможно, что аутоантитела, направленные против NC1, нарушают функцию этих адгезивных протеинов так, что якорные фибриллы коллагена полноценно не взаимодействуют с ними, а связываются с другими соединительнотканными компонентами зоны базальной мембраны и сосочкового слоя дермы. Такое изменение, т.е. потеря функции якорных фибрилл, ведет к нарушению дермо-эпидермального соединения.
В последующем было продемонстрировано, что некоторые аутоантитела у больных, страдающих приобретенным буллезным эпидермолизом, могут иметь мишенью не только NC1 домен, но и NC2 домен со спиральным (центральным) коллагеновым доменом. Это позволяет предположить, что последние также могут играть роль в поддержании функциональной полноценности коллагена VII типа и якорных фибрилл.
Ряд авторов считает, что большинство аутоантител в сыворотке крови пациентов относятся как к комплемент-активируемым, так и комплемент-неактивируемым субклассам IgG. При этом факт присутствия или отсутствия комплемент-активирующих аутоантител субклассов IgG в крови, титр которых может достигать 1:640, не коррелирует с клиническими формами заболевания. Это позволяет предположить, что аутоантитела могут иметь разную природу, в равной степени способствующие формированию пузырей как при комплемент-зависимом воспалительном повреждении, так и комплемент-независимом процессе, сопровождающимся механическим разрывом якорных фибрилл коллагена VII типа. Участие системы комплемента приводит к более выраженным клиническим проявлениям воспалительного приобретенного буллезного эпидермолиза, который клинически сходен с буллезным пемфигоидом и рубцующимся пемфигоидом.
По мнению ряда авторов в некоторых случаях патогенетическая роль при указанной патологии принадлежит циркулирующим аутоантителам класса IgA или одновременно IgA и IgG. С помощью непрямого метода иммунофлюресценции в сыворотке крови больных выявляют антитела IgG и/или IgA к антигенам базальной мембраны эпидермиса, а именно к антигенам lamina densa (рис. 18, b).
В ходе иммуноморфологического исследования биоптатов кожи больных приобретенным буллезным эпидермолизом первоначально были выявлены отложения IgG в зоне базальной мембраны эпидермиса, сходные с таковыми при буллезном пемфигоиде. В дальнейшем было обнаружено, что при буллезном пемфигоиде депозиты IgG располагаются в пределах полудесмосом и lamina lucida, а при приобретенном буллезном эпидермолизе - ниже, в пределах lamina densa и sublamina densa (иммуноэлектронная микроскопия). Прямым методом иммунофлюоресценции в криостатных срезах клинически непораженных участков кожи больных приобретенным буллезным эпидермолизом отмечается выраженная фиксация IgG в виде линейных отложений в зоне базальной мембраны эпидермиса, а в месте формирования пузыря - на его дне (lamina densa), что является диагностическим признаком данного буллезного дерматоза (рис. 18, c). В той же локализации могут быть обнаружены IgA, IgM, С3-компонент комплемента, но в меньшей степени интенсивности реакции.
Итак, приобретенный буллезный эпидермолиз - редкое аутоиммунное заболевание кожи и слизистых оболочек, который помимо классического течения может протекать атипично и имитировать другие буллезные дерматозы, такие как буллезный пемфигоид, рубцующийся пемфигоид или линейный IgA-зависимый дерматоз. В связи с этим имеются сложности в постановке клинического диагноза. Дифференцировать этот буллезный дерматоз позволяют только результаты иммунологических и иммуноморфологических исследований.
Рисунок 15. Больная приобретенным буллезным эпидермолизом (классический вариант).
a - кожа предплечья: ближе к области локтевого сустава располагается пузырь с прозрачным содержимым, плотной покрышкой на слегка гиперемированном фоне;
b - кожа нижних конечностей: около коленных суставов на месте травм располагаются эрозивные дефекты с ярко-красным дном, в области коленного сустава - множественные милиумы, расположенные группами на слегка гиперемированном фоне (справа);
c - слизистая языка: на боковой поверхности языка симметрично располагаются спавшиеся пузыри с плотной покрышкой белесоватого цвета, на кончике языка - пузырек размером до 0,5 см в диаметре с плотной покрышкой и серозным содержимым;
d - кожа кистей: на тыльной поверхности кисти располагаются в местах травм эрозивные дефекты, ногтевые пластины утолщены, отслаиваются.
Рисунок 16. Приобретенный буллезный эпидермолиз (воспалительный вариант).
c - кожа предплечья: буллы с твердой покрышкой (частично спавшиеся) и прозрачным содержимым, расположенные на гиперемированном фоне;
d - кожа бедра: пузырь с твердой покрышкой и прозрачным содержимым, расположенный на слегка гиперемированном фоне, множественные эрозивные дефекты различных размеров на месте вскрывшихся пузырей.
Рисунок 17. Больная приобретенным буллезным эпидермолизом паранеопластического генеза.
a - тотальное поражение кожи с образованием обширных эрозивных поверхностей на месте пузырей, заживление которых происходит с формированием стойкой гиперпигментации;
Рисунок 18. Материалы больных приобретенным буллезным эпидермолизом. х 400.
a - криостатный срез биоптата кожи больной приобретенным буллезным эпидермолизом. Участок клинически интактной кожи. Окраска гематоксилином и эозином. Начальная стадия формирования подэпидермального пузыря - щель между эпидермисом и дермой (стрелка справа) и подэпидермальный пузырь (стрелка слева);
b - криостатный срез кожи донора здорового человека, обработанный сывороткой больной приобретенным буллезным эпидермолизом. Непрямой метод иммунофлюоресценции. Фиксация IgG в зоне базальной мембраны эпидермиса, в месте обычного для заболевания расслоения эпидермиса от дермы;
c - криостатный срез кожи больного приобретенным буллезным эпидермолизом. Прямой метод иммунофлюоресценции. Характерная фиксация IgG на дне (стрелка) подэпидермального пузыря (lamina densa).
Новосибирский государственный медицинский университет
Новосибирский государственный медицинский университет
Санкт-Петербургская государственная медицинская академия им. И.И. Мечникова
Врожденный буллезный эпидермолиз
Журнал: Архив патологии. 2018;80(4): 54-60
Новосибирский государственный медицинский университет



Новосибирский государственный медицинский университет
Новосибирский государственный медицинский университет
Санкт-Петербургская государственная медицинская академия им. И.И. Мечникова
В настоящее время патогенетически обоснованное лечение не выработано и исследование патологии продолжается в аспекте поиска новых маркеров и генов-кандидатов с надеждой на разработку в перспективе таргетной и генной терапии [4]. Надежным способом установления точного диагноза является проведение генетического анализа, кроме биопсии кожи с трансмиссионной электронной микроскопией и/или иммунофлюоресценцией.
Классификация
Тяжесть БЭ высоковариабельна — от локализованных форм с минимальным воздействием на качество жизни до быстро прогрессирующих летальных. При генерализованных тяжелых формах хронический характер поражений, а также вовлечение слизистых оболочек могут привести к системным осложнениям: хроническому воспалению с болевым синдромом и истощением, амилоидозу, контрактурам и даже плоскоклеточной карциноме. Некоторые специфичные формы БЭ ассоциированы с другими кожными проявлениями: поражением ногтей, алопецией, гиперпигментацией, пальмоплантарной кератодермой или внекожными патологиями, такими как мышечная дистрофия или атрезия пилоруса [2, 4].
Несмотря на полиморфизм клинической картины, БЭ делят на три основные формы в зависимости от уровня поражения эпидермиса и дермы, возникающего вслед за механической травмой кожи: простая (epidermolysis bullosa simplex), пограничная (junctional БЭ) и дистрофическая форма (dystrophic БЭ), относительно недавно в группу БЭ внесен синдром Киндлера, характеризующийся дезинтеграцией на разных уровнях эпидермиса [2, 5].
В соответствии с ресурсом OMIM (Online Mendelian Inheritance in Man) [6] и по последнему консенсусу по БЭ [3] простая форма БЭ определяется мутациями в генах кератинов 5 и 14 (KRT5 и KRT14) и делится на четыре основных подтипа с аутосомно-доминантным наследованием: 1) локализованный, ранее известный как БЭ Вебера—Коккейна (Weber—Cockayne (OMIM 131800), с возможностью дополнительной мутации в гене интегрина В4 (ITGB4); 2) генерализованный средней тяжести, ранее известный как Б.Э. Кебнера (Koebner, (OMIM 131900); 3) с пестрой пигментацией (OMIM 131960) и доминированием мутации в гене кератина 5 (KRT5) и 4) тяжелый генерализованный подтип, или БЭ Доулинга—Меара (Dowling—Meara, OMIM 131760).
Пограничная форма БЭ наследуется аутосомно-рецессивно, характеризуется мутациями в трех генах, кодирующих гликопротеины ламинины А3, С2 и В3 (LAMA3, LAMC2 и LAMB3 соответственно), а также мутациями в генах коллагена 17-го типа (COL17A1) и интегрина α6β4, и делится на два основных подтипа: 1) летальный Б.Э. Герлитца (Herlitz, OMIM 226700) и 2) не-Герлитца (OMIM 226650) (табл.1).
В отличие от простой и пограничной формы дистрофическая форма БЭ гетерогенна по типу наследования: 1) аутосомно-доминантная форма (DDEB, OMIM 131750); 2) аутосомно-рецессивный тяжелый генерализованный БЭ Аллопо—Сименса (RDEB, OMIM 226600) и 3) рецессивный дистрофический БЭ инверсный, который не внесен в базу OMIM. Важно отметить, что у 95% больных с дистрофической формой БЭ находят также мутации в гене коллагена 7-го типа (COL7A1). Уровень дезинтеграции – под плотной пластинкой базальной мембраны эпидермиса (sublamina densa).
Патогенез и диагностика БЭ
По новому определению C. Jackson и соавт. [7], БЭ — тяжелое заболевание с преимущественным поражением кожи, обусловленное мутациями как минимум в одном из генов, кодирующих структурные компоненты контактов базальных эпидермоцитов между собой и их базальной мембраной.

Два десятилетия назад выделили более 20 различных форм врожденного БЭ, каждая отличалась по клиническим особенностям, внекожным проявлениям, характеру наследования и локализации кожного дефекта. В последние годы в свете новых научных знаний о молекулярных и генетических аномалиях достигнут значительный прогресс в понимании механизмов, ответственных за формирование Б.Э. Эпидермодермальный контакт является результатом сложных молекулярных взаимодействий между базальными кератиноцитами и компонентами эпителиальной базальной мембраны, в том числе реализуемых с помощью гемидесмосом и якорных фибрилл. Выявление генетических мутаций, вызывающих различные формы БЭ, лежит в основе молекулярной классификации (по дефицитным белкам) (табл. 1) Таблица 1. Классификация врожденного буллезного эпидермолиза по дефицитным белкам (С. Chiaverini и соавт., 2016) [3, 5].
Микроскопически для БЭ характерно субэпидермальное или внутриэпидермальное расположение пузырей, гистологическое исследование часто не позволяет различить локализацию пузырей по отношению к базальной мембране.
Клинический диагноз верифицируют иммуногистохимическим исследованием биоптата кожи с определением уровня пузыря и дефицитного белка. Гистологическая картина врожденного БЭ близка БЭ приобретенному (acquisita) — хроническому заболеванию, связанному с аутоиммунной реакцией на коллаген 7-го типа в субэпидермальных анкерных структурах фибрилл [8]. Исследование биоптата кожи не позволяет дифференцировать БЭ acquisita и буллезный пемфигоид, необходимо проведение прямой иммунофлюоресценции. В обзоре [9] представлены современные представления об аутоиммунных буллезных дерматозах.
Электронно-микроскопическое исследование (ЭМИ) позволяет точно определить локализацию пузырей и более подробно изучить морфологические изменения при различных вариантах болезни. Этот метод в течение многих лет сохранял ведущие позиции среди методов диагностики буллезных дерматозов для подтверждения диагноза основных типов Б.Э. Начиная с первого применения ЭМ для исследования БЭ этот метод остается наиболее точным для определения уровня дезинтеграции эпидермиса, что лежит в основе современной классификации БЭ и близких к нему патологических процессов [10, 11]. Другие методы исследования, в том числе иммунофлюоресценция и анализ ДНК, приобретают все большее значение в диагностике, тем не менее ни один метод до сих пор не может полностью вытеснить ЭМИ, хотя оно в настоящее время используется в основном в экспертных лабораториях и редко применяется в качестве первичной диагностической процедуры.
По сравнению с другими методами диагностики БЭ ЭМИ имеет как сильные, так и слабые стороны. Во-первых, при сравнительно легких формах БЭ результаты иммунофлюоресценции могут не отличаться от нормы, в то время как ЭМИ позволяет идентифицировать микротрещины и другие ультраструктурные аномалии. Тем не менее нарушения могут быть весьма незначительными, что требует обширного опыта в интерпретации ультраструктурных изменений. ЭМИ имеет большое значение для диагностики БЭ Доулинга—Меара (Dowling—Meara) особенно у новорожденных до возникновения типичных герпетиформных волдырей. Иммунофлюоресценция может не выявить аномалии отчасти из-за интраэпидермальных трещин, но ЭМИ способно продемонстрировать характерные изменения тонофиламентов в базальных клетках [12].
Наряду с этим ЭМИ полезно для диагностики врожденной буллезной ихтиозиформной эритродермии (или эпидермолитического гиперкератоза), что позволяет выявить комки тонофиламентов, расположенные в верхних слоях эпидермиса.
По сравнению с другими лабораторными методами, в частности такими, как рутинная гистопатология, ЭМИ является очень дорогостоящей процедурой, требует специального оборудования и трудоемкой высококвалифицированной пробоподготовки. Строгое соблюдение ключевых принципов получения образца помогает избежать ненужной траты времени, усилий и затрат на получение и обработку биопсии кожи для диагностики с помощью ЭMИ. Все образцы кожи необходимо тщательно исследовать, и патогномоничные изменения часто можно видеть лишь в одном из образцов [15].
Биопсия кожи от сформированных более 1 ч пузырей или старых повреждений, как правило, дают противоречивые результаты, связанные с некрозом и регенерацией эпидермиса и дермы. Поэтому рекомендуется сепарация эпидермиса с помощью осторожного растирания кожи в месте предполагаемой биопсии. В идеале материал для биопсии должен содержать участок границы нового поражения, чтобы были видны в микроскоп и расслоения, и сохраненные участки кожи. В чрезвычайно хрупкой коже, например при Б.Э. Герлитца (Herlitz), взятие биопсии кожи часто бывает достаточно, чтобы вызвать отслоение эпидермиса от дермы. Напротив, в некоторых подтипах БЭ, в частности при БЭ Вебера—Коккейна, индукция пузыря затруднена даже при локальном тепловом воздействии и обширном растирании кожи, так что диагностическая польза биопсии является, как правило, сомнительной. Кроме того, в большинстве случаев простой БЭ имеет семейный анамнез и соответствующие клинические признаки, достаточные для подтверждения диагноза. Предпочтительнее не брать биопсию, если все соответствующие последующие шаги не будут заранее учтены, доступны и достижимы [12].
Электронно-микроскопические особенности основных типов БЭ

При всех подтипах простого БЭ дезинтеграция имеется в нижней части эпидермиса, как правило, под ядрами базальных клеток, тогда как при поверхностных БЭ уровень формирования пузыря находится на границе между зернистым и роговым слоем эпидермиса (табл. 2) Таблица 2. Электронно-микроскопические особенности врожденного буллезного эпидермолиза [16].
При БЭ Доулинга—Меара (Dowling—Meara) вдобавок к внутриэпидермальному пузырю описаны аномальные кластеры тонофиламентов (кератиновые промежуточные филаменты) в основном в базальных кератиноцитах [5, 17]. Эта ультраструктурная особенность является патогномоничной, дающей ключи к пониманию имеющейся молекулярной аномалии.
При подтипе БЭ с дефицитом плектина, также известном как БЭ с мышечной дистрофией, расщепление локализуется сразу над уровнем гемидесмосом, которые часто сравнительно миниатюрны по размеру [18]. Подобный тип расщепления также характерен для БЭ с атрезией пилоруса, который трудно предвидеть, поскольку этот вариант БЭ обычно считается подтипом пограничного БЭ [19]. Кроме того, редкий не-Герлитца пограничный БЭ также обусловлен мутациями в гене коллагена 17-го типа и ассоциирован с внутриэпидермальным расщеплением [18].
Наконец, при редкой форме генерализованного аутосомно-рецессивного простого БЭ, вызываемого null-мутацией в гене кератина 14, сеть кератиновых филаментов в базальных эпидермоцитах сильно скомпрометирована и нормальные тонофиламенты немногочисленны или полностью отсутствуют [20].
Пограничный (Junctional) буллезный эпидермолиз
Особенностями пограничного Б.Э. Герлитца, подозреваемого обычно при прозрачном пузыре на уровне lamina lucida, являются мелкие или редуцированные гемидесмосомы, однако в ряде случаев находят дезинтеграцию на уровне базальных отделов базальных клеток, обусловленную, наиболее вероятно, нарушением молекулярных связей кератиновых филаментов с гемидесмосомами [21]. Эти структурные особенности также видны при БЭ с атрезией пилоруса [19].
В случаях не-Герлитца пограничного БЭ, возникающего в результате мутаций в ламинине-5 (ламинин-332) или коллагене 17, гемидесмосомы бывают как мелкие и малочисленные, так и нормальные по размерам и количеству [12, 20]. Сравнительные исследования, демонстрирующие количественные или качественные ультраструктурные различия между этими двумя главными генетическими подтипами не-Герлитца пограничного БЭ, не проводились.
Дистрофический буллезный эпидермолиз
Плоскость расщепления обычно локализована непосредственно под lamina densa. Еще один наиболее полезный диагностический маркер при генерализованной аутосомно-рецессивной (или Аллопо—Сименса (Hallopeau—Siemens) форме — это отсутствие нормальных якорных фибрилл. При доминантной и более локализованной рецессивной форме якорные фибриллы присутствуют, но в меньшем количестве, чем в норме. Детальный количественный анализ не выявил различий якорных фибрилл между этими разными генетическими подтипами. Расстройство, известное как переходный БЭ новорожденного, характеризуется очевидным замедлением секреции коллагена 7 и наличием связанных с мембраной включений, содержащих мелкодисперсный или аморфный материал в базальных кератиноцитах [5], который был иммуногистохимически определен как коллаген 7-го типа [22].
Электронно-микроскопические особенности эпидермиса и дермы при патологических процессах, относительно недавно классифицированных как формы БЭ
Ключом к пониманию патогенеза эктодермальной дисплазии с истончением кожи стало генетически обусловленное нарушение структуры десмосом, вызванное мутацией гена плакофилина-1. Заболевание характеризуется выраженной сепарацией эпидермоцитов и расширением межклеточных пространств, ассоциированными с миниатюрными десмосомами в супрабазальных слоях [21, 24]. Кроме того, выражена перинуклеарная конденсация кератиновых филаментов, и лишь небольшое их количество локализуется на периферии клеток, участвуя в организации десмосом. Дезинтеграция клеток, как представляется, вызвана атрофией пальцеподобных цитоплазматических выростов, направленных к десмосомальным контактам. Аналогичные ультраструктурные особенности описаны в случае летального акантолитического БЭ simplex, обусловленного делецией в хвостовом домене десмоплакина [25].
Ларинго-онихо-кожный синдром — редкое заболевание, вызванное N-терминальной делецией в альфа-3a-изоформе ламинина. Пациенты редко имеют истинные волдыри, но страдают от весьма характерных эрозий на лице и в других местах. С помощью электронной микроскопии продемонстрировано фокальное расширение lamina lucida в сочетании с мелкими гемидесмосомами [26].
Таким образом, ЭМИ чрезвычайно важно для диагностики пузырных дерматозов, электронная микроскопия предоставила маркеры для классификации как самих дерматозов, так и процессов с аналогичными клиническими проявлениями. Ультраструктурные исследования внесли неоценимый вклад в поиск генетических основ различных типов дерматозов с нарушениями молекулярной организации десмосом [12]. Кроме того, результаты ЭМИ важны при молекулярно-биологических исследованиях, нацеленных на разработку новых методов лечения, направленных на эпителизацию поражений при рецессивных дистрофических пузырных дерматозах [27].
Наличие системности проявлений, гетерогенность клинических форм, сходство множества ультраструктурных проявлений патологии эпидермодермального контакта при БЭ и акантолитической пузырчатке [15], а также экспрессия антигена пемфигоида 1 при обеих патологиях свидетельствуют о сходстве патоморфогенеза буллезных дерматозов в целом. Основные отличия эпидермолизов и пузырчатки связаны с более ранней манифестацией эпидермолизов (при рождении или сразу после него) и наличием антител к антигену пемфигоида 1 при пузырчатке. Именно поэтому надежды на разработку подходов к лечению связаны с универсальными молекулами, экспрессируемыми структурными компонентами эпидермиса и дермы.
Лечение и перспективы

Перспективные таргеты для лечения врожденного БЭ, а также ряда буллезных дерматозов — антигены эпидермальной базальной мембраны представлены на схеме эпидермодермального контакта (см. рисунок). Схема эпидермодермального сопряжения и компонентов эпителиальной базальной мембраны с акцентом на локализации антигенов, являющихся перспективными таргетами для лечения БЭ (помечены желтым). Использованы элементы рисунка из [31, стр. 143]. Кроме того, ведутся интенсивные исследования в области протеиновой, генной или клеточной заместительной терапии, аллогенной трансплантации костного мозга, использования пуповинной крови или трансплантации плюрипотентных стволовых клеток.
Взятые у истощенного и умирающего от БЭ 7-летнего пациента клетки были культивированы и модифицированы с помощью методов генной терапии, что позволило ex vivo получить полноценный эпидермис, который был имплантирован с помощью 3 операций и в течение 2 лет функционирует без рецидива. Это исследование демонстрирует высокие достижения структурной биологии ХХI века и открывает перспективы для реального внедрения методов клеточной и генной терапии в клиническую практику.
Читайте также: