
Ангидрозная эктодермальная дисплазия наследуется как сцепленный с х хромосомой
Обновлено: 28.05.2024
Ангидротическая эктодермальная дисплазия – наследственное заболевание, обусловленное генетическим дефектом и характеризующееся врожденными аномалиями развития зубов, потовых и сальных желез, волосяных фолликулов и желез слизистых оболочек.
Триада, специфичная для данной патологии:
- ан- или гипогидроз (снижение активности потовых желез);
- гипо-, олиго- или адонтия (дефекты зубного ряда);
- гипотрихоз (скудость волосяного покрова).
Женщины могут быть носителями дефектного гена (клинически значимых проявлений заболевания в этом случае нет, симптомы минимальны: нарушение развития отдельных зубов, скудное оволосение и пониженная потливость), болеют исключительно лица мужского пола. Полный симптомокомплекс заболевания у женщин является парадоксальным, встречается крайне редко.
Впервые заболевание было описано в 1848 году французским ученым Дж. Туреном; в 1913 году немецким стоматологом Дж. Кристом и в 1929 году немецким дерматологом Х. Сименсом были детализированы клинические проявления.
В настоящее время точная частота встречаемости патологии не установлена, предположительно – у одного новорожденного на 5000–10 000. Заболевание выявляется на всех континентах, у представителей всех рас в равной степени.
Синонимы: синдром Криста – Сименса – Турена, гипогидротическая эктодермальная дисплазия.
Внимание! Фотография шокирующего содержания.
Для просмотра нажмите на ссылку.
Причины и факторы риска
В настоящее время локализованы три гена на разных хромосомах, которые могут являться причиной развития названного заболевания. Известно количество мутаций в указанных генах, на настоящий момент их насчитывается более 200:
- ген EDA, кодирующий белок эктодисплазин-А, входит в состав хромосомы Хq12‑1;
- ген EDAR, кодирующий рецептор фактора некроза опухоли (член суперсемейства EDAR), картирован на хромосоме 2q11‑q13;
- ген TDARADD, кодирующий эктодисплазин-А рецептор ассоциированный белок-адаптор, хромосома 1q42.2‑
Предполагают, что действие генетического дефекта проявляется на 2–5 месяце внутриутробного развития.

Синдром эктодермальной дисплазии передается аутосомно-рецессивным путем
Тип наследования – Х-сцепленный, рецессивный: в данном случае патологический ген локализован в половой Х-хромосоме и у женщин подавляется доминантным геном, сцепленным с другой Х-хромосомой. Поскольку мужчины являются носителем только одной Х-хромосомы, в которой располагается генетический дефект, и дублирование генов в такой ситуации невозможно, развивается развернутая клиническая картина с многообразием симптомов.
Женщины могут быть носителями дефектного гена, но болеют исключительно лица мужского пола. Полный симптомокомплекс заболевания у женщин является парадоксальным, встречается крайне редко.
Симптомы
Клинические проявления заболевания обусловлены некорректным формированием наружного зародышевого листка (эктодермы) во внутриутробном периоде (структуры, формирующиеся из эктодермального зародышевого листка, включают зубы, эпидермис и его дериваты, нервную систему и органы чувств):

Для эктодермальной дисплазии характерна коническая форма передних зубов с острым режущим краем
Диагностика
Постановка корректного диагноза возможна при всесторонней оценке клинической картины и данных лабораторных и инструментальных исследований, генетической экспертизы:
- общий анализ крови (отмечается снижение цветного показателя, количества гемоглобина – менее 100-90 г/л);
- биохимический анализ крови (выявляются уменьшение содержания белковых фракций альбуминов и увеличение гамма-фракций, снижение содержания кальция, фосфора, железа);
- гистологическое исследование биоптата кожи (анализируется состояние потовых желез);
- исследование волос (устанавливаются уменьшение диаметра стержня волоса, изменение его формы от круглой до уплощенной, атрофия или отсутствие коркового слоя);
- исследование генов EDA, EDAR, TDARADD на предмет мутаций;
- пренатальная (дородовая) ДНК-диагностика.
Для ангидротической эктодермальной дисплазии характерна следующая триада: снижение активности потовых желез, дефекты зубного ряда, скудость волосяного покрова.
Лечение
Специфического лечения не существует, заболевание пожизненное.
Возможные осложнения и последствия
Осложнениями ангидротической эктодермальной дисплазии могут быть:
- присоединение вторичных инфекций вследствие снижения иммунитета;
- заболевания ЖКТ (гастрит, дуоденит, язвенная болезнь);
- заболевания дыхательной системы (бронхиальная астма, хронический бронхит, синусит);
- заболевания органов зрения (кератит, конъюнктивит, блефарит);
- воспалительные заболевания кожи;
- аллергические заболевания.
В настоящее время точная частота встречаемости патологии не установлена, предположительно – у одного новорожденного на 5000–10 000.
Прогноз
При своевременной диагностике, незначительной степени выраженности клинических проявлений и адекватных методах профилактики осложнений прогноз благоприятный.
Прогноз значительно ухудшается при множественности симптомов, поздней диагностике и отсутствии адекватного ухода.
Об авторе

Информация является обобщенной и предоставляется в ознакомительных целях. При первых признаках болезни обратитесь к врачу. Самолечение опасно для здоровья!
Стоматологи появились относительно недавно. Еще в 19 веке вырывать больные зубы входило в обязанности обычного парикмахера.
Упав с осла, вы с большей вероятностью свернете себе шею, чем упав с лошади. Только не пытайтесь опровергнуть это утверждение.
Человеческие кости крепче бетона в четыре раза.
В течение жизни среднестатистический человек вырабатывает ни много ни мало два больших бассейна слюны.
У 5% пациентов антидепрессант Кломипрамин вызывает оргазм.
Даже если сердце человека не бьется, то он все равно может жить в течение долгого промежутка времени, что и продемонстрировал нам норвежский рыбак Ян Ревсдал. Его "мотор" остановился на 4 часа после того как рыбак заблудился и заснул в снегу.
Самая высокая температура тела была зафиксирована у Уилли Джонса (США), который поступил в больницу с температурой 46,5°C.
Если улыбаться всего два раза в день – можно понизить кровяное давление и снизить риск возникновения инфарктов и инсультов.
Многие наркотики изначально продвигались на рынке, как лекарства. Героин, например, изначально был выведен на рынок как лекарство от детского кашля. А кокаин рекомендовался врачами в качестве анестезии и как средство повышающее выносливость.
Печень – это самый тяжелый орган в нашем теле. Ее средний вес составляет 1,5 кг.
Согласно исследованиям, женщины, выпивающие несколько стаканов пива или вина в неделю, имеют повышенный риск заболеть раком груди.
Согласно исследованиям ВОЗ ежедневный получасовой разговор по мобильному телефону увеличивает вероятность развития опухоли мозга на 40%.
Каждый человек имеет не только уникальные отпечатки пальцев, но и языка.
В Великобритании есть закон, согласно которому хирург может отказаться делать пациенту операцию, если он курит или имеет избыточный вес. Человек должен отказаться от вредных привычек, и тогда, возможно, ему не потребуется оперативное вмешательство.

Прошлое, настоящее и будущее проблемы установления отцовства
Задача точного установления отцовства — это такая же древняя проблема, как и поиски смысла жизни. Во все времена мужчин интересовало, своих ли они растят детей.
Лариса Алексеевна Сафонова в своем докладе затронула одну из актуальных проблем современной медицины. По ее словам, в настоящее время большое внимание уделяется медицине будущего, или 4П-медицине.
Предиктивная (to predict (с англ.) - предсказывать) — способная предсказывать развитие болезни еще до клинических проявлений.
Персонализированная (to personalize (с англ.) - персонализировать) — данный аспект в большей части относится к фармакотерапии и назначении медикаментозного лечения с учетом генотипа.
Превентивная (to prevent (с англ.) - предупреждать) — направленная на предупреждение заболевания.
Партисипативная (to perticipate (с англ.) - участвовать) — пациент является активным участником собственного лечения, его информируют и обучают, помогают в выборе.
4П-медицина базируется на достижениях биомедицинской науки последних лет, в особенности на генетике. Предполагается, что генетика станет основой профилактики и лечения заболеваний.
Некоторые заболевания волос имеют генетическую основу и экогенетическую сверхсоставляющую. Для того, чтобы определить генетическую основу заболевания имеются различные методы генетических исследований. И наиболее полномасштабный метод исследования полногеномный поиск ассоциаций.
Полногеномный поиск ассоциаций
Полногеномный поиск ассоциаций (GWAS – genome – wide – association study) – исследование ассоциаций между геномными вариантами и различными фенотипическими проявлениями. Анализ ассоциаций генов-кандидатов лежит в основе идентификации генетических вариантов предрасположенности.
Известно около 90 наследуемых заболеваний и синдромов, сопровождающихся алопецией или гипотрихозом. В большинстве случаев врожденный гипотрихоз является одним из признаков эктодермальных дисплазий, изолированных или сочетанных. В тех случаях, когда изменения волос доминируют и имеют большое диагностическое значение, их рассматривают как отдельное заболевание.
Затем Сафонова Л.А. подробнее рассмотрела некоторые заболевания волосистой части головы.
Монилетрикс
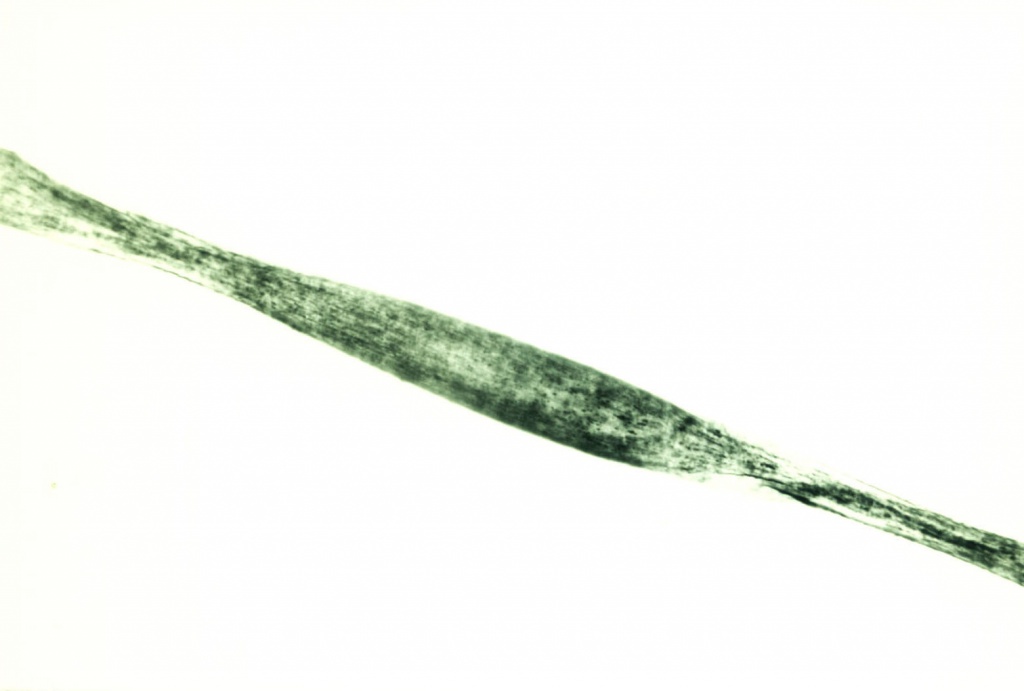 | 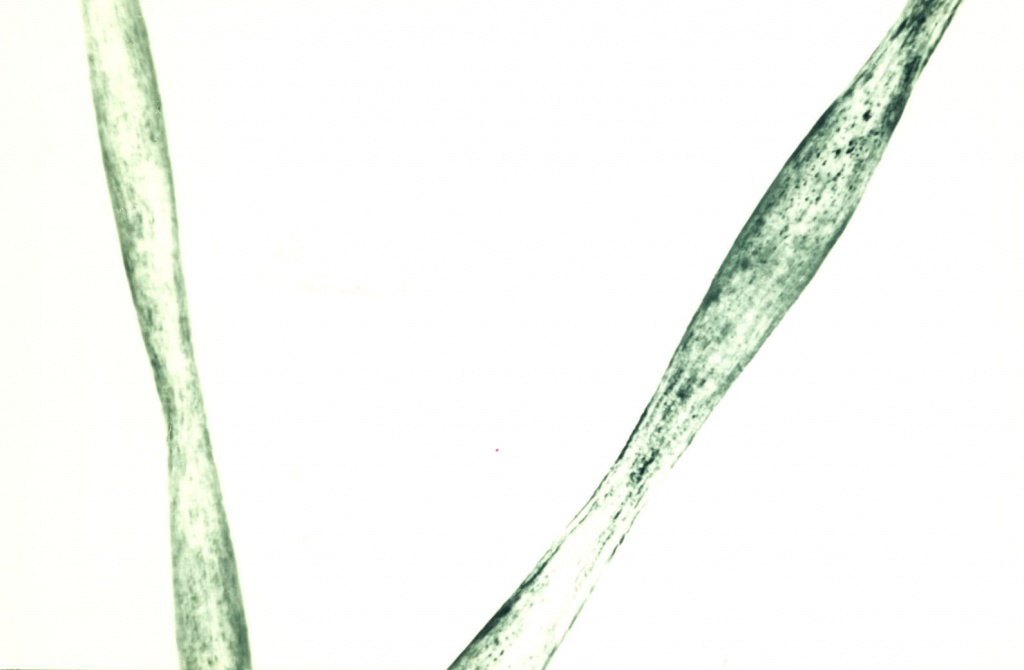 |
Это заболевание генетически гетерогенно. В большинстве случаев наследуется по аутосомно-доминантному типу. У мальчиков заболевание встречается чаще, чем у девочек.
Клинические признаки заболевания:
гипотрихоз, повышенная ломкость и поредение волос
нарушение ороговения волосяных фолликулов с образованием фолликулярных гиперкератотических узелков.
На трихоскопии: чередование перетяжек веретенообразных вздутий на стержнях волос.
как правило, любое лечение малоэффективно
назначают поливитаминные препараты
кратковременно, до уменьшения выраженности фолликулярного кератоза, назначают витамин А в дозах около 100 000 ЕД или неотигазон в дозе 0,5 мг/кг.
Синдром Нетертона
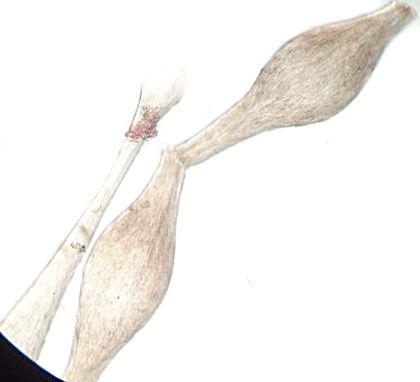 |  |
Наследуется аутосомно-рецессивно. Обнаружены мутации в гене SPINKS 5q32 первичным продуктом которого является фермент-ингибитор серин-протеазы Kazal type 5, который в норме определяется в эпителиальной и лимфоидной тканях.
Предполагается нарушение формирования липидного барьера кожи, а именно его ламеллярной субстанции, что проявляется снижением резистентности к бактериальным патогенам и повышенной склонностью к воспалительным процессам.
Клинические признаки заболевания:
гипотрихоз. повышенная ломкость волос
ихтиозиформные изменения кожи
симптоматическое. Новорожденные не редко нуждаются в интенсивной терапии, лечении системными антибиотиками.
наружно назначают слабые кератолитические средства, мази и кремы с кортикостероидами, ретиноидами.
системные ретиноиды следует применять с осторожностью ,у ряда больных на их фоне возможно обострение процесса.
Врожденный наследственный гипотрихоз
Заболевание наследуется по аутосомно-доминантному типу. Происходит миниатюризация волос и замена терминальных волос пушковыми. Встречается редко.
Клиническое проявление заболевания:
волосы с рождения редкие, легко обламываются.
в раннем детстве становятся более густыми, но примерно с пубертатного возраста практически полностью выпадают сначала в теменной области, а затем и по краям.
характерна высокая лобная и затылочная линия волос. Может развиваться тотальная алопеция особенно у мужчин
зубы и ногти не изменены
в зоне поражения могут быть признаки перифолликулярного рубцевания
с рождения выявляются милиарные кисты
примерно у 50% больных выявляется увеличенная щель между центральными резцами верхней челюсти (диастема).
возможно сочетание заболевания с атопическим дерматитом, синдромом Черногубова-Элерса-Данлоса, наследственной ювенильной дистрофией сетчатки.
Трихотиодистрофия
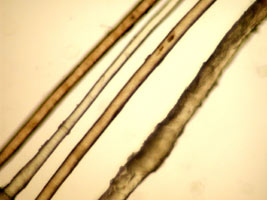 |
Трихотиодистрофия представляет собой заболевание из группы нейроэктодерманых дисплазий. Развитие болезни объясняют дефицитом серы.
Однако, уровень серосодержащих аминокислот в крови в пределах нормы, что исключает алиментарный этиологический фактор.
Наследуется по аутосомно-рецессивному типу.
Клинические проявления заболевания:
гипотрихоз, повышенная сухость и ломкость волос
структурные нарушение стержней волос
снижение содержания серосодержащих аминокислот в стержнях волос на 10-50%
ихтиозиформные изменения кожи
задержка умственного и физического развития, преждевременное старение
врожденная двусторонняя катаракта
Диагноз ставят на основании клинической картины, данных микроскопии волос, анализа микроэлементарного состава волос.
Синдром Менкеса
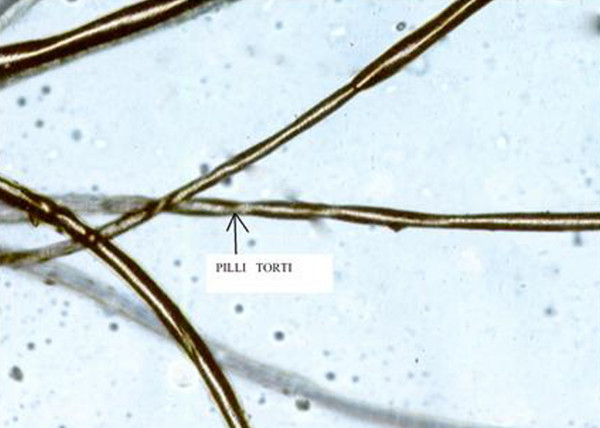 | 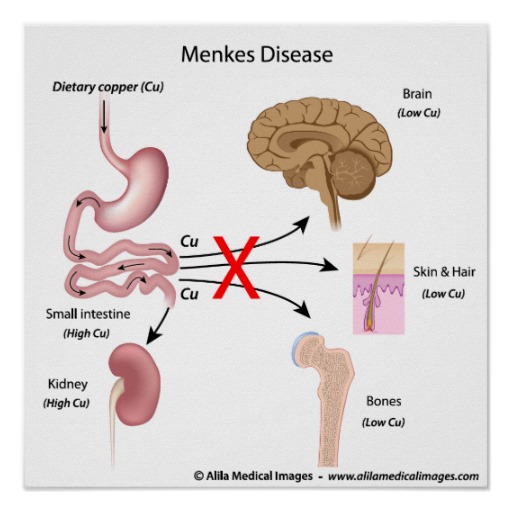 |
Развитие синдрома связано с дефицитом меди вследствие нарушения ее абсорбции. Заболевание наследуется рецессивно, сцеплено с Х-хромосомой. Нарушается работа АТР фермента, обеспечивающего доставки CU2+. Значительное снижение содержание меди отмечается в сыворотке крови, ткани мозга и печени. В плазме резко снижено содержание церулоплазмина. В то же время высокие концентрации Сu2+ обнаруживают в тканях пищевода, почек, легких, селезенки, поджелудочной железы, коже и мышцах. В основном болеют мальчики.
Клинические проявления заболевания:
структурные аномалии стержня волос, гипотрихоз
тяжелая умственная и физическая отсталость
задержка роста и патология костной ткани
высокая подверженность инфекциям
аномалии почек и сосудов, зрительные расстройства
диагноз ставят на основании клинической картины, данных световой микроскопии волос, биохимического анализа крови
Важно своевременное распознавание болезни, так как парентеральное введения препарата Cu2+ дает обнадеживающие результаты только при очень раннем их назначении — в течение первого месяца жизни.
При уже возникших изменениях в ЦНС клинического эффекта не наблюдается, даже при нормализации уровня Cu2+ в плазме крови.
Синдром неукладываемых волос
Синдром неукладываемых волос имеет аутосомно-доминантный тип наследования. В основе изменений формы волосяного стержня лежит нарушение кератинизации и изменение конфигурации внутреннего корневого влагалища.
Клинические проявления заболевания:
волосы светлые, с серебристым оттенком
они довольно густые, скорость их роста не замедлена, повышенной ломкости нет
после расчесывания волосам не удается придать желаемую форму
иногда наблюдается слабо выраженные дефекты зубной эмали и ногтевых пластин
при более тяжелом течении синдром может сочетаться с пигментной дистрофией сетчатки, катарактой, олигодонтией, брахидактилией
Диагноз устанавливается на основании клинической и гистологической картины, а также характерных ультраструктурных признаков (на поверхности волоса определяется продольное углубление, а поперечный срез имеет вид почки или треугольника, иногда - уплощенного элипса.
Гнездная алопеция (ГА)
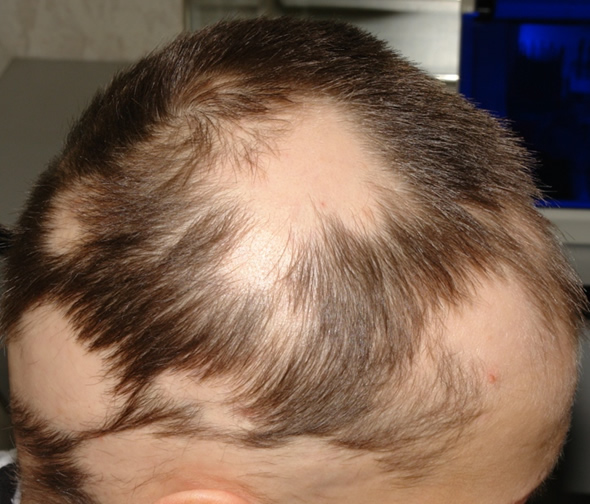 | 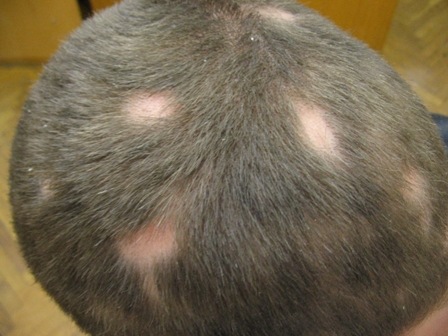 |
ГА — хронический мультифактериальный дерматоз, обусловленный аутоиммунным поражением клеток волосяного фолликула, развивающийся на фоне генетической предрасположенности и характеризующийся образованием очагов выпадения волос и частым поражением ногтевых пластин.
Гены, связанные с ГА
Angela Christiano et al. (2010) обнаружили корреляцию между геном ULBP3 на 6 хромосоме в локусе q25.I и ГА. В норме этот ген не выделяется, но при ГА ULBP3 белки обнаруживаются в большой концентрации в ВФ. ULBP3 соединяется с рецепторами NK — клеток, называемых NKG2D, вызывая перифолликулярное воспаление. ULBP3 гены могут быть использованы в качестве биомаркеров при ГА.
Выделяют и другие гены, которые некоторые ученые связывают проявлениями ГА.
Так исследователи указывают на ген EDA2R, мутации в котором приводят к потере волос.
Выявлены гены, которые участвуют в потере волос и влияют на структуру волос. Одним из этих генов является ген P2RY5. При мутации в данном гене в волосяном фолликуле происходят определенные биохимические процессы, приводящие к сухости, ломкости и поредению волос.
В 2009 году в Японии был обнаружен ген SOX21 , который помогает регулировать процесс синтеза кератина и, таким образом, является ведущим регулятором формирования волос.
Андрогенетическая алопеция (АГА)
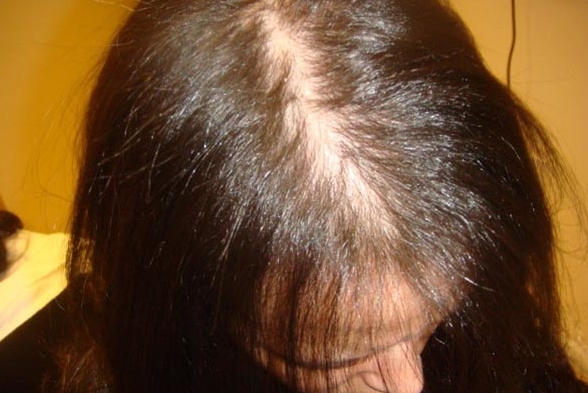 |  |
АГА — поредение волос, возникающее в результате генетически обусловленной повышенной чувствительности волосяных фолликулов к мужским гормонам. Выявлена ассоциация АГА и с ишемической болезнью сердца, артериальной гипертензией, ожирением, раком простаты и др.
Механизм развития АГА и гены
Тип наследования - полигенный. Андрогенный ген AR, влияющий на мужское облысение, наследуется по линии хромосомы Х, передаваемой по материнской линии и находится в положении Хq12. Второй ген расположен на 20й хромосоме 20р11.22. Третий — 3q26, расположен на 3й хромосоме. Второй и третий гены могут быть унаследованы как от отца, так и от матери.
Ген AR расположенный на Х-хромосоме в положении Хq12 является рецептором андрогенов. Его функцией является метаболизм стероидных гормонов. Андрогеновый рецептор является внутриклеточным фактором транскрипции, который активизируется путем связывания с тестостероном. При связывании с гормоном, рецептор перемещается в ядро и связывается со специфичной последовательностью молекулы ДНК. Связанный комплекс тестостерон — AR активирует или подавляет экспрессию тестостерон-регулирующих белков. Основные — полиморфизмы — CAG повторы в 1 экзоне.
Значение исследования CAG повторов гена AR в общей практике
Исследования полиморфизма CAG-повторов в 1 экзоне гена андрогенетического рецептора (AR) имеет диагностическое и прогностическое значение, позволяющее определить риск репродуктивных нарушений, рака простаты, установить причину мужского бесплодия, мышечной атрофии.
Диапазон 20 — 26 повторов считается относительной нормой. При уменьшенном количестве CAG-повторов ( 26) свидетельствует о наличии генетической предрасположенности к гормонозависимому нарушению сперматогенеза.
Увеличение числа CAG-повторов до 38 — 62 приводит к спинобульбарной мышечной атрофии типа Кеннеди.
Результаты генетического теста позволяют врача внедрять предсимптоматическую оценку риска, что позволяет провести раннюю диагностику и предотвратить наступление заболевания, предложив персонализированные рекомендации. Синтез традиционных методов обследования и результатов генетического тестирования позволяет персонифицировать рекомендации по применению лечебных мероприятий и их длительность.
Идеальный инструмент для пренатальных исследований. Уникальное качество изображения и весь спектр диагностических программ для экспертной оценки здоровья женщины.
Введение
Синдром ЕЕС (ectrodactyly-ectodermal dysplasia-clefting syndrome) - редкий генетический синдром, обычно проявляющийся триадой признаков: расщелиной лица, эктродактилией конечностей и признаками эктодермальной дисплазии [1]. Выделяют 2 типа синдрома: ЕЕС-1 и ЕЕС-3. Ранее был описан также и ЕЕС-2, однако на сегодняшний день локализация гена, кодирующего ЕЕС-2, считается ошибочной, следовательно, и формы ЕЕС-2 не существует. Первый тип характеризуется мутацией в 7-й хромосоме в области 7q11.2-q21.3, третий - имеет в большинстве случаев мутацию Тp63-гена и другие новые, спонтанные мутации на 3-й хромосоме 3q27 [2]. В OMIM (ежедневно обновляемом международном классификаторе Online Mendelian Inheritance in Man), где представлены все известные на сегодняшний день фенотипы и генотипы менделирующих (наследуемых) болезней человека, эти виды синдрома кодируются по-разному: ЕЕС-1 - 129900, а ЕЕС-3 - 604292 [3].
ЕЕС-синдром - наследственная патология с аутосомно-доминантным типом наследования, характеризующаяся различной пенетрантностью (проявляемостью) и экспрессивностью (степенью выраженности) [1].
Манифестные признаки синдрома ЕЕС (синдромальное ядро)
Эктродактилия - это расщелина кистей и/или стоп с отсутствием одного или нескольких центральных пальцев, известная также под названием "Split hand-split foot malformation" (SHFM). Часто внешний вид кистей и стоп сравнивают с клешней краба. Нередко встречается в сочетании с синдактилией (сращением пальцев).
Эктодермальная дисплазия проявляется особенностями строения всех производных эктодермы. Изменения затрагивают волосы, зубы, ногти [11]. Волосы и ресницы у таких людей тусклые, редкие и жесткие. Характерны аномалии зубов, гипоплазия зубной эмали, снижение пигментации волос, кожи, радужной оболочки глаз, что вызывает светлый их цвет. Также часто сочетание синдрома с фотобоязнью [1], обструкцией носо-слезных каналов [12], кондуктив- ной тугоухостью [13].
Эктодермальная дисплазия проявляется отсутствием потовых желез, что характеризуется наличием гипогидротинового синдрома, проявляющегося в резкой сухости и шелушении кожи. Характерным проявлением этого синдрома является хриплый, осипший голос из-за нарушения увлажнения голосовых связок, вследствие чего не происходит полного их смыкания [14].
Нарушение речи у больных с ЕЕС-синдромом имеет много причин. Во-первых, это связано с последствиями наличия расщелины губы и/или неба и "гиперназальной" речью, во-вторых, правильной артикуляции может препятствовать не только изменение голосовых связок, но и патология зубов. В-третьих, из-за аномалий слуховых косточек часто прогрессирует тугоухость, ведущая к когнитивным расстройствам и расстройствам речи.
Дети с этим синдромом обычно имеют нормальный уровень интеллекта. Лечение основано на коррекции пороков лица и конечностей. Признаки эктодермальной дисплазии проявляются в постнатальном периоде [1]. Врожденные пороки развития (ВПР) мочевыделительной системы у больных с синдромом ЕЕС возникают более чем в половине случаев. Среди них описаны: мегауретер, гидронефроз, уретероцеле, аплазия почки, аномалии гениталий, уретровезикальный рефлюкс с частыми инфекциями мочевыделительных путей [12].
Пренатальная диагностика синдрома ЕЕС
Учитывая выраженность проявлений при синдроме ЕЕС, пренатальная диагностика этой патологии, безусловно, возможна. Однако на сегодняшний день в мировой литературе встречается незначительное число публикаций, посвященных диагностике этого редкого синдрома 17, что связано, видимо, с редкостью возникновения данной аномалии. В основном сроками постановки диагноза являются 16-30 недель беременности. Большую помощь в диагностике данной патологии, учитывая выраженные изменения фенотипа лица и конечностей, оказывает применение новых ультразвуковых технологий 3D/4D с методиками поверхностной реконструкции [16].
Первое описание пренатальной диагностики синдрома датировано 1993 годом и принадлежит M. Bronshtein и R. Gershoni, когда при применении трансвагинальной эхографии у плода была выявлена расщелина губы и неба и эктродактилия в 14 недель беременности [17]. Уникальность этого факта в том, что, пожалуй, впервые из известных случаев диагностики врожденных аномалий дебют дородового выявления порока принадлежит сроку первого триместра. Примечательно и то, что авторами не только описаны отдельно выявленные пороки развития плода, но и пренатально установлена нозология этого состояния. Другими словами, диагноз не звучал, как "множественные врожденные пороки развития", когда невозможно говорить об этиологии заболевания, а следовательно, и о мерах специфической профилактики данной патологии в дальнейшем в семье. Пренатальный диагноз был выставлен полно и абсолютно корректно, что особенно важно при проведении медико-генетического консультирования с формированием тактики адекватного репродуктивного поведения семьи в дальнейшем.
Из-за незначительного количества публикаций, посвященных пренатальной диагностике этого генетического синдрома, представляем ряд собственных наблюдений диагностики синдрома ЕЕС в разные сроки беременности, в том числе и в первом триместре. Особо рассмотрим особенности проведения медико-генетического консультирования при диагностике различных форм синдрома с аутосомно-доминантным типом наследования для выработки специфических мер профилактики данного наследственного заболевания.
Клиническое наблюдение 1
Пациентка Н. 24 лет. Беременность первая. Женщина и муж соматически здоровы, брак неродственный. Обратилась в медико-генетическое отделение МОНИИАГ в 21,4 недели беременности в связи с подозрением на порок развития конечностей у плода. Были выявлены: эктродактилия кистей (рис. 1) и стоп плода (рис. 2.). Из особенностей строения выявлены двусторонние пиелоэктазии (рис. 3). Проведено медико-генетическое консультирование. Семья приняла решение прервать данную беременность в связи с наличием инвалидизирующих пороков конечностей. При патологоанатомическом исследовании ВПР конечностей подтверждены, а также дополнительно выявлена расщелина мягкого неба и признаки эктодермальной дисплазии (гипертрофия десен, характерный лицевой фенотип). Окончательный диагноз: "синдром ЕЕС, аутосомно-доминантный тип наследования". Мутация de novo.

7. Мужчина, страдающий диабетом и косолапостью, вступает в брак со здоровой (по этим признакам) женщиной. Рождаются дети: первый – диабетик, но не косолапый; второй – косолапый, но не диабетик. Определить вероятность того, что такие дети в этой семье могли появиться, если известно, что косолапость и отсутствие наследственно обусловленной предрасположенности к диабету наследуются по аутосомно-доминантному типу.
Ответ. Детей с диабетом, без косолапости – 1/4; косолапых, без диабета – 1/4.
2.2. Тригибридное скрещивание
1. У матери ребенка – ll группа крови, круглое лицо, тонкие кости; у отца – lll группа крови, продолговатое лицо, нормальная толщина костей. Вычислить, какова вероятность появления в данной семье ребенка с l группой крови, внешне похожего на отца (продолговатое лицо, нормальная толщина костей), если известно, что гены, контролирующие формирование костей нормальной толщины и продолговатый овал лица, – рецессивные и расположены в разных парах аутосом.
Решение. Заносим данные задачи в таблицы 6 и 7, составляем схему 6.
Таблица 7. Группы крови по системе АВО
В данном случае, очевидно, целесообразно составить три самостоятельные схемы скрещивания (схемы 7–9), рассмотреть сначала отдельно вероятность появления ребенка с каждым интересующим нас признаком, а затем, перемножив все три полученные вероятности, прийти к окончательному ответу.
Окончательный ответ: 1/4 ґ 1/2 ґ 1/2 = 1/16.
Следует отметить, что использование таких букв латинского алфавита, как D/d и Е/е, объясняется тем, что две первые буквы – А/а и B/b – оказались в этой задаче уже использованными при рассмотрении передачи по наследству от родителей к потомству групп крови (система АВО, схема 10).
ПРИЗНАКИ, СЦЕПЛЕННЫЕ С ПОЛОМ
Признаки, за развитие которых отвечают гены, локализованные в Х-хромосоме
1. Доминантные гены, локализованные в X-хромосоме
1. Женщина, имеющая гипоплазию (истончение) эмали, выходит замуж за мужчину, у которого такой же дефект. От этого брака рождается мальчик, не страдающий данной болезнью. Какова была вероятность появления в этой семье здорового мальчика? Какова вероятность появления в этой семье здоровой девочки? Известно, что ген, ответственный за развитие гипоплазии эмали, – доминантный ген, локализованный в X-хромосоме.
Составим решетку Пеннета (табл. 2), проанализируем ее содержание и получим единственно верный ответ.
Нетрудно догадаться, что появление в этой семье первенца, здорового мальчика (относительно рассматриваемого заболевания), имеющего генотип ХaY, говорит о том, что генотип больной матери этого ребенка был гетерозиготным, т.е. ХAХa, что и представлено нами как в схеме скрещивания, так и в решетке Пеннета.
При анализе данных решетки Пеннета становится ясно, что вероятность появления здорового мальчика была равна в этой семье 25% (от всех детей). Вероятность же появления в этой семье здоровых девочек абсолютно исключена.
2. От брака мужчины, у которого нет рахита, устойчивого к лечению витамином D, и женщины, страдающей этим заболеванием, рождается здоровая, как и ее отец, девочка. Может ли данная семья быть абсолютно уверенной в том, что и все последующие дети, родившиеся в этой семье, будут такими же здоровыми, как и эта девочка? Известно, что ген, ответственный за развитие этой болезни, – доминантный ген полного доминирования, локализованный в X-хромосоме.
Появление в семье здоровой девочки с генотипом XаXа свидетельствует о том, что ее мать является носительницей рецессивного гена, т.е. имеет генотип XAXа. Изобразим это на схеме (схема 2).
Построим решетку Пеннета (табл. 4) и рассчитаем, какой шанс был у этой девочки родиться здоровой, а также определим, насколько велика вероятность того, что и все последующие дети, которые могут появиться в этой семье, будут здоровыми (относительно данного заболевания).
Из данных решетки Пеннета видно, что вероятность рождения в этой семье здоровой (без рахита) дочери была равна 25% (от всех детей). Вообще же вероятность рождения в данной семье здоровых и больных детей (как среди девочек, так и среди мальчиков) равна 1:1.
То есть 50% девочек (от всех девочек), так же, как и 50% мальчиков (от всех мальчиков), будут здоровы; вторая половина детей этой семьи будет больна рахитом.
2. Рецессивные гены, локализованные в Х-хромосоме
1. В семье молодых здоровых родителей, не подверженных частым инфекционным заболеваниям (пневмония, отиты и др.), рождаются три девочки-погодки. Можно ли считать, что и они, и все последующие дочери в этой семье в дальнейшем будут такими же устойчивыми к бактериальным инфекционным заболеваниям, как и их родители, если известно, что бабушка этих детей по материнской линии и дедушка по отцовской линии имеют очень хрупкое здоровье (ввиду того, что у них так называемая болезнь Брутона, т.е. врожденный недостаток Y-глобулинов, что и обуславливает склонность к определенным инфекционным заболеваниям). Ген, ответственный за развитие состояния дефицита глобулинов, – рецессивный ген, локализованный в X-хромосоме.
При анализе вероятности того, какими (относительно рассматриваемого признака) будут девочки, родившиеся в этой семье, нужно учесть следующее
1. Так как бабушка этих девочек (по материнской линии) была больна, то ее генотип – это наверняка рецессивная гомозигота, т.е. ХaХa и, следовательно, все 100% ее гамет – это Xa.
3. Теперь об отце девочек, у которого, согласно условию задачи, рассматриваемого заболевания – болезни Брутона – нет. Следовательно, его генотип может быть представлен как ХAY.
То, что дедушка девочек (по отцовской линии) был болен, никак не может отразиться на генотипе отца девочек. Ведь он получил от своего отца Y-хромосому, которая не несет генов, ответственных за наличие или отсутствие этой болезни.
Теперь уместно построить решетку Пеннета (табл. 6).
Анализ данных решетки Пеннета позволяет сделать такой вывод: в этой семье все три девочки, несомненно, будут здоровыми; болезнь Брутона, имевшаяся у представителей их рода, не проявит себя ни у одной из них.
2. У женщины, страдающей отсутствием потоотделения (ангидрозная эктодермальная дисплазия), и мужчины, не имеющего указанного дефекта, рождается сын. Определить, унаследует ли ребенок болезнь матери или же мальчик будет здоровым, как и его отец. Известно, что ген, ответственный за развитие этой болезни, – рецессивный ген, локализованный в X-хромосоме. будет ли страдать этим заболеванием девочка, являющаяся вторым ребенком в семье?
Как видно из анализа решетки Пеннета, и мальчик, который уже родился, и все остальные мальчики, которые могут появиться в этой семье, непременно будут страдать нарушением потоотделения. Напротив, 100% потомков женского пола, которые могут появиться в этой семье, будут характеризоваться наличием нормального потоотделения.
3. Известно, что ген гемофилии (несвертываемость крови) – рецессивный ген, локализованный в X-хромосоме. Здоровая женщина, мать которой так же, как и она, была здоровой, а отец страдал гемофилией, вышла замуж за мужчину, страдающего гемофилией. Появление какого потомства можно ожидать от этого брака (относительно рассматриваемого заболевания)? Использовать при решении этой задачи такую весьма распространенную форму изображения половых хромосом: X-хромосома – как тире (—); Y-хромосома – как полустрелка (—).
При построении решетки Пеннета (табл. 9) следует записывать гаметы и зиготы с учетом введенной символики.
Анализируя полученные результаты, можно сделать следующие выводы.
1. Все девочки, родившиеся в этой семье, здоровы, но имеют гетерозиготный генотип: (1/4 от всех зачатых детей).
2. Появление здоровых мальчиков и мальчиков-гемофиликов можно ожидать с равной вероятностью (по 1/4 от всех зачатых детей).
3. Наконец, при комбинации (1/4 от общего количества детей) должна бы родиться девочка, но этого, увы, не происходит, так как ген гемофилии справедливо считается полулетальным геном, т.е. в случае возникновения рецессивной гомозиготы мы имеем летальный исход.
Женщины-гемофилики в природе не существуют!
3. Признаки, за развитие которых отвечают гены, локализованные в Y-хромосоме
1. Ген, ответственный за развитие такого признака, как гипертрихоз (оволосение края мочки уха), – один из немногих рецессивных генов, локализованных в Y-хромосоме. Если мужчина с гипертрихозом женится на женщине, у которой, естественно, гипертрихоза нет, то каков реальный шанс появления в этой семье детей с гипертрихозом, как мальчиков, так и девочек?
Составляем схему скрещивания 6.
Как говорится, комментарии излишни: все 100% девочек, рожденных от этого мужчины, здоровы; все мальчики – с гипертрихозом.
Решение. Рецессивный ген синдактилии, локализованный в Y-хромосоме, несомненно, в 100% случаев передается от отца к сыну. Казалось бы, ни схема скрещивания, ни решетка Пеннета здесь не требуются вовсе.
И тем не менее если избрать такую форму обозначения половых хромосом, какой мы однажды пользовались, то ради любопытства можно проделать работу, ставшую уже довольно-таки привычной для тех, кто привык (и полюбил!) решать задачи по генетике (табл. 11, схема 7).
Читайте также: