
Синдром поланда берут ли в армию
Обновлено: 02.07.2024
Деформации грудной клетки встречаются у 2% людей. Изменения (дефекты) в костных и хрящевых тканях снижают как опорную функцию грудной клетки, так и необходимый объем подвижности. Деформации грудной клетки (грудины и ребер) являются не только косметическим дефектом и вызывают не только психологические проблемы, но и довольно часто приводят к нарушению функции органов грудной клетки (сердечно-сосудистой системы и дыхательной системы).
Причины
Причины деформации грудной клетки могут быть как врожденными, так и приобретенными. Основные причины следующие:
- Кифоз
- Хронические обструктивные заболевания легких
- Синдром Марфана
- Аномалии остеогенеза
- Ахондроплазия
- Синдром Тернера
- Синдром Дауна
- Эмфизема
- Рахит
- Врожденные аномалии ребра
- Астма
- Неполное сращение грудины плода
- Врожденное отсутствие грудной мышцы
- Болезнь Бехтерева
- Воспалительный артрит
- Остеомаляция
В клинической практике чаще всего встречаются воронкообразная деформация грудной клетки и килевидная деформация.
Воронкообразная деформация грудной клетки (впалая грудь)
Воронкообразная деформация грудной клетки (впалая грудь) на сегодняшний день является наиболее распространенной деформацией грудной клетки и встречается в 1 случае из 400 новорожденных. Килевидная деформация, как вторая наиболее распространенная форма деформации, встречается в 5 раз реже, чем воронкообразная грудная клетка.
Этиология развития воронкообразной деформации
Существует несколько теорий, объясняющих развитие этой деформации, но до конца этиология остается неясной. Некоторые авторы считают, что развитие воронкообразной деформации может быть связано с чрезмерно быстрым ростом реберного хряща, который вытесняет грудину кзади. Аномалии диафрагмы, рахит, или повышенное внутриматочное давление также предположительно способствуют смещению задней части грудины. Частое ассоциация воронкообразной деформации с другими заболеваниями опорно-двигательного аппарата, такими как синдром Марфана, дает возможность предполагать, что в определенной степени деформации обусловлены аномалиями соединительной ткани. Генетическая детерминированность встречается также у 40% пациентов с килевидной деформацией.
Клинические проявления
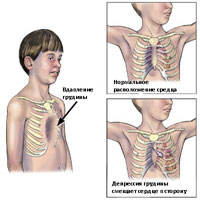
Воронкообразная грудная клетка может проявляться как в виде небольшого дефекта, так и выраженного дефекта, при котором грудина доходит почти до позвонков. Возникновения дефекта является результатом 2 факторов: (1) степенью задней ангуляции грудины и степенью задней ангуляции реберного хряща в зоне прикрепления ребер к грудине. Если же, кроме того имеются дополнительно ассиметрии грудины или хрящевые ассиметрии, то в таком случае оперативное лечение становится более технически сложным.
Воронкообразная деформация возникает, как правило, при рождении или вскоре после рождения. Деформация часто прогрессирует, и глубина вдавления увеличивается по мере роста ребенка. Впалая грудь чаще встречается у мужчин, чем у женщин, в соотношении 6:1 Впалая грудь может сочетаться с другими врожденные аномалиями, включая аномалии диафрагмы. У 2% пациентов, впалая грудь связана с врожденными аномалиями сердца. У пациентов с характерным габитусом тела, можно предположить диагноз синдром Марфана.
Существует несколько методов количественной оценки тяжести деформации при воронкообразной груди, которые обычно включает измерения расстояния от грудины к позвоночнику. Возможно, наиболее часто используемым методом является метод Халлера, который использует отношение поперечного расстояния до переднезаднего расстояния, полученные на основании КТ. В системе Haller, оценка 3,25 или выше свидетельствует о тяжелом дефекте, который требует хирургического вмешательства.
Воронкообразная грудь вообще не оказывает особого физиологического воздействия на младенцев или детей. Некоторые дети испытывают боль в области грудины или реберного хряща, особенно после интенсивных нагрузок. У других детей возможно сердцебиение, что может быть связано с пролапсом митрального клапана, который обычно имеет место у пациентов со впалой грудью. Некоторые пациенты могут чувствовать шум движения крови, который связан с тем, что легочная артерия находится близко к грудине и во время систолы пациент может отмечать шум выброса крови.
Иногда у пациентов с воронкообразной грудью встречается астма, но отмечено что деформация не оказывает явного влияния на клиническое течение астмы. Воронкообразная деформация оказывает влияние на сердечно-сосудистую систему и наблюдения показали, что после оперативной коррекции деформации происходит значительное улучшение функций сердечно-сосудистой системы.
Килевидная деформация
Килевидная деформация является второй наиболее распространенной врожденной деформацией грудной стенки. Pectus carinatum составляет примерно 7% всех деформаций передней грудной стенки. Она чаще встречается у мальчиков, чем у девочек (соотношение 4:1). Как правило, эта деформация имеется уже при рождении и имеет тенденцию к прогрессированию по мере роста ребенка. Килевидная деформация представляет собой выпячивание грудной клетки и фактически представляет собой спектр деформаций, которые включают костохондральный хрящ и грудину. Изменения в костнохондральном хряще могут быть как односторонними, так и двухсторонними. Кроме того, выпирание грудины может быть как большим, так и незначительным. Дефект может быть асимметричным, вызывая ротацию грудины с депрессией с одной стороны и выпиранием с другой стороны.
Этиология
Патогенез килевидной деформации, также как и воронкообразной деформации не ясен. Высказывалось предположение, что это результат избыточного разрастания ребер или остеохондральных хрящей. Существует определенная генетическая детерминированность килевидной деформации. Так в 26% случаев отмечено наличие семейного анамнеза этой формы деформации. Кроме того в 15 % случаев килевидная деформация сочетается со сколиозом, врожденными пороками сердца, синдромом Марфана или другими заболеваниями соединительной ткани.
Клинические проявления
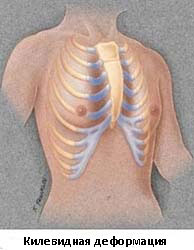
Килевидную деформацию можно разделить на 3 различных типа деформаций.
- Тип 1. Xарактеризуется симметричным выступом грудины и реберных хрящей. При этом типе деформации грудины мечевидный отросток смещены вниз
- Тип 2. Корпорокостальный тип, при этом типе деформации происходит смещение грудины вниз и вперед или выгибание средней или нижней трети грудной клетки. Этот тип деформации, как правило, сопровождается искривлением ребер.
- Тип 3. Костальный тип. При этом типе деформации задействованы в основном реберные хрящи, которые выгибаются вперед. Искривления грудины, как правило, не значительны.
Симптомы при килевидной деформации чаще встречаются у подростков и могут быть в виде сильной одышки, возникающей при минимальной нагрузке, снижение выносливости и появление астмы. Происходит это вследствие того, что экскурсия стенки грудной клетки ограничена из-за фиксированного переднезаднего диаметра грудной клетки, что приводит к увеличению остаточного объема, тахипноэ, и компенсационной диафрагмальной экскурсии.
Синдром Поланда
Синдром Поланда назван в честь Альберта Поланда, который впервые описал этот вид деформации грудной клетки в результате наблюдений в школе и относится к спектру заболеваний, которые связаны с недоразвитием грудной стенки. Этот синдром включает аномалии развития большой грудной, малой грудной мышц, передней зубчатой мышцы, ребер, и мягких тканей. Кроме того, может наблюдаться деформация руки и кисти.
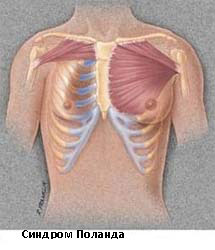
Заболеваемость синдромом Поланда составляет примерно 1 случай на 32 000 родившихся детей. Этот синдром в 3 раза чаще встречается у мальчиков, чем у девочек, и у 75% пациентов поражается правая сторона. Существует несколько теорий относительно этиологии этого синдрома, которые включают в себя аномальную миграцию эмбриональной ткани, гипоплазию подключичной артерии или внутриутробной травмы. Тем не менее, ни одна из этих теорий не доказало свою состоятельность. Синдром Поланда редко ассоциирован с другими заболеваниями. У некоторых пациентов с синдромом Поланда встречается лейкемия. Существует определенная ассоциация этого синдрома с синдромом Мебиуса (односторонний или двусторонний паралич лицевого нерва, отводящего глазного нерва).
Симптомы синдрома Поланда зависят от степени дефекта и в большинстве случаев это косметические жалобы. У пациентов с наличием значительных костными дефектов, могут быть выбухания легкого, особенно при кашле или плаче. У некоторых пациентов возможны функциональные нарушения и дыхательные нарушения. Легкие сами по себе не страдают при этом синдроме. У пациентов со значительными дефектами мышечной и мягких тканей могут стать очевидными снижение толерантности к физическим нагрузкам.
Синдром Жена
Синдром Жена или прогрессирующая дистрофия грудной клетки, которая обусловлена внутриутробным нарушением роста грудной клетки и гипоплазией легких. Этот синдром был впервые описан в 1954 году Женом у новорожденных. И хотя в большинстве случаев такие пациенты не выживают, но в некоторых случаях оперативные методы лечения позволяют таким пациентам жить. Синдром Жена наследуется по аутосомно-рецессивному типу и не было отмечено наличие ассоциации с другими хромосомными нарушениями.
Дефекты грудины
Дефекты грудины можно разделить на 4 типа и все являются редкими: грудная эктопия сердца, шейная эктопия сердца, торакоабдоминальная эктопия сердца и расщепление грудины. Торакальная эктопия сердца представляет собой аномалию расположения сердца вне грудной клетки, и сердце совершенно не защищено плотными костными тканями. Выживаемость пациентов с грудной эктопия сердца очень низкая.Описано только три удачных случая оперативного лечения из 29 операций с этой аномалией.
Шейная эктопия сердца отличается от грудной только локализацией аномального расположения сердца. Как правило, такие пациенты не имеют шансов на выживаемость. У пациентов с торакоабдоминальной эктопией сердце расположены книзу грудины. Сердце покрыто мембраной или тонкой кожи. Смещение сердца вниз является результатом полулунного дефекта перикарда и дефекта диафрагмы. Нередко также бывают дефекты брюшной стенки.
Расщелина грудины является наименее серьезной из 4 аномалий, потому что сердце почти закрыто и находится в нормальном положении. Поверх сердца имеется частичное или полное расщепление грудины, причем частичное разделение встречается чаще, чем полное расщепление. Ассоциации с пороками сердца при этой аномалии встречаются достаточно редко. У большинства детей, расщепление грудины обычно не вызывает особо заметных симптомов. В отдельных случаях, возможны респираторные симптомы в результате парадоксального движения дефекта грудины. Основным показанием для проведения хирургического лечения является необходимость обеспечить защиту сердца.
Диагностика
Диагностика деформаций грудной клетки, как правило, не представляет больших трудностей. На первом плане из инструментальных методов исследования стоит рентгенография, которая позволяет оценить как форму деформации, так и ее степень. КТ грудной клетки позволяет определить не только костные дефекты и степень деформации грудины, но и наличие смещения средостения, сердца, наличие сдавления легкого. МРТ позволяет получить более расширенную информацию, как о состоянии костных тканей, так и мягких тканей и, кроме того, не обладает ионизирующей радиацией.
Функциональные исследования деятельности сердца и легких, такие как ЭКГ, ЭХО- кардиография, спирография позволяют оценить наличие функциональных нарушений и динамику изменений после оперативного вмешательства.
Лабораторные методы исследования назначаются в случае необходимости дифференциации с другими возможными состояниями.
Лечение
При деформации средней и тяжелой степени только оперативное лечение может восстановить нормальную функцию органов грудной клетки.
Использование материалов допускается при указании активной гиперссылки на постоянную страницу статьи.
Главный военный клинический госпиталь внутренних войск МВД России, Балашиха, Московская область
Главный военный клинический госпиталь внутренних войск МВД России, Балашиха, Московская область
Главный военный клинический госпиталь внутренних войск МВД России, Балашиха, Московская область
Главный военный клинический госпиталь внутренних войск МВД России, Балашиха, Московская область
Два случая склеродермии у военнослужащих (возвращаясь к напечатанному)
Главный военный клинический госпиталь внутренних войск МВД России, Балашиха, Московская область



На основании данных литературы рассматриваются атипичные формы (течение) ограниченной склеродермии, особенности классификации и диагностики заболевания. Приводятся два собственных наблюдения склеродермии у военнослужащих, представляющие интерес в плане диагностики и принятия экспертного решения.
Главный военный клинический госпиталь внутренних войск МВД России, Балашиха, Московская область
Главный военный клинический госпиталь внутренних войск МВД России, Балашиха, Московская область
Главный военный клинический госпиталь внутренних войск МВД России, Балашиха, Московская область
Главный военный клинический госпиталь внутренних войск МВД России, Балашиха, Московская область
Увеличение числа больных склеродермией отмечают ряд авторов [1]. Мы не можем об этом судить из-за редкости патологии в воинском коллективе, о чем писали недавно [2]. Вместе с тем за последний год мы встретились с 2 случаями склеродермии со своеобразной клинической картиной, вызвавшей трудности в диагностике и принятии экспертного решения.
Локализованная (ограниченная) склеродермия (ОСД) — хроническое заболевание соединительной ткани, характеризующееся очаговыми воспалительно-склеротическими изменениями кожи и подлежащих тканей без вовлечения в патологический процесс внутренних органов [4].
Общепринятой классификации нет. В зависимости от клинических проявлений большинство авторов, в том числе военные дерматологи [5], различают несколько форм ОСД — бляшечную (пузырную и глубокую), линейную (полосовидную), пятнистую — поверхностную ограниченную (болезнь белых пятен, белый лихен Цумбуша, первичный склероатрофический лихен), идиопатическую атрофодермию Пазини—Пьерини, ОСД с гемиатрофией лица Ромберга. Различные формы ОСД существенно различаются как по ряду клинических и морфологических признаков, так и по течению [1, 4—8].
Идиопатическая атрофодермия Пазини—Пьерини многими авторами считается абортивным (первичным атрофическим) вариантом ОСД и клинически проявляется длительно существующими, незначительно западающими очагами коричневого или серо-коричневого цвета с фиолетово-сиреневым оттенком без признаков уплотнения кожи. Очаги располагаются чаще всего на туловище (спине) и верхних конечностях [4—8].
Некоторые авторы считают склероатрофический лихен Цумбуша и идиопатическую атрофию Пазии—Пьерини самостоятельными заболеваниями.
Алгоритм обследования больного склеродермией основывается в основном на анамнестических данных и физикальном обследовании. В начале развития ОСД, когда уплотнение еще не выражено и имеется только обесцвеченное пятно, процесс может напоминать витилиго или депигментированное пятно при недифференцированной лепре [8]. Обязательные и дополнительные лабораторные исследования указаны во всех современных руководствах [4].
Среди больных преобладают лица с группой крови 0(I), что косвенно подтверждает генетическую обусловленность склеродермии. Несомненна связь склеродермии с состоянием эндокринной системы. Наличие эндокринных расстройств у больных склеродермией является показанием к назначению препаратов гипофиза, щитовидной железы, околощитовидных желез, половых гормонов [9].
Типична эозинофилия. Реакция на антиядерные антитела обычно положительная у пациентов с генерализованным заболеванием. Исследование биопсийного материала позволяет выявить различную степень воспаления (типично для ранних очагов) и склероз дермы [10]. Приводим два собственные наблюдения.
Из анамнеза: со слов, в 12—14-летнем возрасте появилось темное пятно в области поясницы слева. Врачи в военкомате интересовались происхождением пятна, но поскольку жалоб больной не предъявлял, диагноз не выставлялся. Закончил ВВУЗ и служит на командной должности. В 2004 г. появилось коричневое пятно на левой половине туловища; в 2007 г. — два белых пятна на левой боковой поверхности туловища и спине. Пятно в области поясницы постепенно увеличивалось в размере, уплотнения на очагах никогда не замечал.
Результаты специальных исследований: клинический анализ крови и мочи без патологии; группа крови Аβ (II) резус-положительная; RW — отрицательная, антитела к ВИЧ, НВS антиген, anti HCY не обнаружены; ревматоидный фактор, С-реактивный белок, титр АСТЛ-О не обнаружены; антиядерные антитела (полуколичественный анализ) 0,5; антитела к двуспиральной ДНК — 6,5; антитела к односпиральной ДНК — 7,5 (количественное исследование). Биохимия крови без патологии, кроме увеличения уровня непрямого билирубина до 16,4 мкмоль/л; общего до — 17,9 мкмоль/л; АлАТ до 89,4 ед/л.
Рентгенография органов грудной клетки — без патологии. ЭФГДС: недостаточность кардии; очаговый поверхностный гастрит (антральный отдел желудка). Заключение УЗИ органов брюшной полости, почек и щитовидной железы: признаки диффузных изменений печени (жировой гепатоз?, реактивные изменения?). Рекомендован контроль УЗИ печени через 6 мес. Заключение специалистов: ЛОР, невропатолог, хирург — здоров. Окулист: сложный дальнозоркий астигматизм, амблиопия слабой степени левого глаза. Гастроэнтеролог: стеатогепатит низкой активности.

Биопсия с очага на грудной клетке (рис. 2.), Рисунок 2. Гистопатологическая картина ОСД у больного Г., 34 лет. Значительное истончение эпидермиса с атрофией сосочков; мономорфный коллагеноз дермы с очаговой лимфоидной инфильтрацией, редукция сальных и потовых придатков кожи. Окраска гематоксилином и эозином, ув. 50. заключение: фиброматоз подкожной клетчатки с умеренным хроническим воспалением.
Дерматологический диагноз: ограниченная склеродермия.
Лечение в течение 2 нед (ксантинола никотинат 15% 2 мл внутримышечно ежедневно; трентал 400 мг 2 раза в сутки; эссенциале форте по 1 капсуле 3 раза в сутки, солкосерил 5% мазь 2 раза в сутки на очаги; фонофорез с пелоидином (гель ПО-КУР) клинических изменений не вызвало. На военно-врачебную комиссию офицер не представлялся. Взят на диспансерное наблюдение.
Больной В., 20 лет, солдат, поступил в июне 2009 г. с жалобами на слегка зудящие высыпания на голенях и в области коленных суставов, появившиеся в марте и увеличившиеся в количестве.

Результаты специальных исследований: клинический анализ крови без патологии, кроме относительной эозинофилии 8—11%; анализ мочи без патологических изменений; группа крови 0 (I) резус-положительная; RW — отрицательная, антитела к ВИЧ, НВS антиген, anti HCY не обнаружены; ревматоидный фактор и С-реактивный белок не обнаружены, титр АСТЛ-О 200 МЕ/мл; антиядерные антитела 1,0; гормоны щитовидной железы: ТЗ свободный — 5,66 пмоль/л; Т4 свободный — 10,28 пмоль/л; ТТГ— 0,75 МЕ/мл; антитела к тирепероксидазе — менее 0,16 МЕ/мл. Инцизионная биопсия с очага на голени (рис. 4, а—в), Рисунок 4. Гистопатологическая картина ОСД у больного В., 20 лет. Истончение эпидермиса, редукция сосочков; поля гиалиноза коллагеновых волокон-пучков, с частичным замещением подкожной клетчатки и участки гомогенизации коллагена дермы; диффузно-очаговая лимфоцитарная инфильтрация (и вокруг сосудов); редукция сальных, потовых, волосяных придатков кожи. Окраска гематоксилином и эозином, ув.100 (а, б); ув. 200 (в). заключение: гистологическая картина не противоречит клиническому диагнозу. Рентгенография органов грудной клетки — без патологии; придаточных пазух носа: недоразвитие лобной пазухи. Снижение пневматизации клеток решетчатого лабиринта. При УЗИ органов брюшной полости и почек патологии не выявлено; паренхима щитовидной железы эхонеоднородна за счет анэхогенных множественных образований размерами до 5 мм. Объем щитовидной железы в пределах возрастной нормы. Заключение: ультразвуковые признаки множественных коллоидных кист щитовидной железы. Эндокринолог: коллоидный зоб, эутиреоз. Дерматологический диагноз: ОСД.
В результате проведенного лечения (за время обследования): курс пенициллина, антигистаминная терапия, эутирокс, физиотерапия — очаги остались прежних размеров, но исчезли границы в виде сиреневого валика, явления атрофии сохраняются (рис. 3, ж).
Решением военно-врачебной комиссии больной признан: В — ограниченно годен к военной службе. Заболевание получено в период военной службы.
У одного больного могут быть представлены различные формы ОСД и даже системной склеродермии, что подтверждает мнение о единстве этих форм заболевания. Очаги могут находиться в различной стадии развития [4—8].
Дифференциальный диагноз атрофодермии Пазини—Пьерини с бляшечной склеродермией представляет лишь академический интерес и основывается на отсутствии мраморно-белых, деревянистой плотности бляшек с сиреневым венчиком и склероза дермы, характерных для склеродермии [6].
Течение ОСД медленное, с прогрессированием склероза кожи. Несмотря на различную клиническую картину в обоих случаях первые проявления ОСД возникли в юношеские годы и в дальнейшем медленно прогрессировали в виде появления новых очагов. У офицера по сравнению с солдатом отмечается более длительный анамнез и по гистологической картине явления фиброза более выражены. Профилактика ОСД заключается в диспансерном наблюдении и лечении для предотвращения рецидивов и утяжеления заболевания.

Синдром Поланда — редкий врожденный синдром, характеризующийся частичным или полным односторонним отсутствием большой грудной мышцы и врожденным пороком развития кисти со стороны поражения грудной клетки. Также могут быть аномалии ребер (аплазия или гипоплазия) и грудины (килевидная или воронкообразная деформация), которые отчетливо пальпируются ввиду истончения подкожно-жирового слоя. Часто при данном синдроме имеются аномалии сосково-ареолярного комплекса. Дефекты грудной клетки обычно носят косметический характер. Мужской пол превалирует над женским в соотношении 2–3 : 1, по частоте преобладает правостороннее поражение. На данный момент общепринятой теорией развития синдрома Поланда является нарушение кровотока по подключичной и/или позвоночной артериям и их ветвям на 6-й неделе внутриутробного развития. Ввиду полиморфности проявлений синдрома существует множество подходов к объему и срокам оперативной коррекции. В статье мы проанализировали данные по этиопатогенезу и клинической картине синдрома Поланда, а также по способам хирургического лечения.
Ключевые слова
Полный текст
Введение
Синдром Поланда — врожденный синдром, характеризующийся чаще односторонней (реже двусторонней) гипоплазией (аплазией) большой грудной мышцы (возможно, и малой грудной мышцы) и ребер (как правило, от 2 до 5), ателией (дефект сосково-ареолярного комплекса), амастией (отсутствие ткани молочной железы), истончением подкожно-жирового слоя грудной клетки, отсутствием оволосения в подмышечной области и на грудной клетке со стороны поражения, а также врожденными пороками развития верхней конечности, чаще — в виде симбрахидактилии.
Различают две формы заболевания: полную и неполную. Сочетание дефектов грудной клетки и кисти называется полным синдромом Поланда, изолированная гипоплазия большой грудной мышцы диагностируется как неполный синдром (встречается значительно чаще, чем полная форма) [1]. Однако, по мнению N. Yiyit et al. (2015), для постановки данного диагноза необходимо сочетание одного или более симптомов в сочетании с поражением большой грудной мышцы.
Частота встречаемости синдрома Поланда составляет 1 случай на 10–100 тысяч человек [2].
Мужской пол превалирует над женским в соотношении 2–3 : 1 [3].
Отдельные симптомы данного заболевания впервые были отмечены L.M. Lallemand (1826) и R. Frorier (1839), но более полное описание данного синдрома дал английский студент-медик Альфред Поланд в 1841 году [4].
В 1895 году Thompson обобщил все имеющиеся аномалии, характерные для данного синдрома, но окончательно свое имя в честь Альфреда Поланда синдром получил в 1962 году в работе Clarkson (цит. по: Shamberger, 1998; Slezak and Sasiadek, 2000).
Этиология и патогенез
Этиология и патогенез заболевания до сих пор точно неизвестны.
На данный момент общепринятой теорией развития синдрома Поланда является нарушение кровотока по подключичной и/или позвоночной артерии и их ветвям на 6-й неделе внутриутробного развития. Тяжесть проявлений синдрома определяется длительностью и интенсивностью нарушения кровообращения в бассейне данных артерий [5].
Большинство случаев синдрома Поланда являются спорадическими, только примерно в 1 % выявляется наследственный фактор. Считается, что данный синдром имеет аутосомно-доминантный тип наследования [6].
Czeizel et al. (1990) обследовали 18 типичных пациентов с синдромом Поланда, из них только у одного больного был отягощен наследственный анамнез: у отца отсутствовала большая грудная мышца слева с тяжелой гипоплазией большого пальца левой кисти, синдактилией II–III пальцев, тогда как у сына имелась умеренная гипоплазия большой грудной мышцы слева с тяжелой гипоплазией большого пальца левой кисти [7].
В 2014 году Vaccari et al. сообщили о случае синдрома Поланда у монозиготных близнецов с делецией 11q12.3 с вовлечением пяти генов, четыре из которых (HRASLS5, RARRES3, HRASLS2 и PLA2G16) ответственны за регуляцию клеточного роста, дифференцировку клеток и механизм апоптоза через ras-сигнальный путь. Нарушение данных механизмов развития может привести, помимо нарушения дифференцировки мышечных клеток и к онкологическим заболеваниям [8].
В 1977 году B. C. McGillivray, R. B. Lowry опубликовали результаты обследования 44 пациентов с данным синдромом, семейный анамнез которых не был отягощен. У всех больных отсутствовала грудинная часть большой грудной мышцы с одной стороны, у 40 был отмечен порок развития верхней конечности и только у 4 человек верхняя конечность была интактна [9].
С 1841 года по настоящее время в мире описано около 500 случаев синдрома Поланда.
Клиническая характеристика
Наиболее подробную клиническую характеристику синдрома Поланда удалось найти в работе N. Yiyit et al. (2015), основанную на результатах обследования 113 больных в возрасте от 6 до 38 лет. Правостороннее поражение авторы отмечали в 55,7 %, левостороннее — в 37,1 %, двустороннее — в 7 % наблюдений. По данным Jones (1926), Lord et al. (1990), правостороннее поражение встречается в 75 % случаев (цит. по: Jones (1926), Lord et al. (1990)).
Как указывалось выше, основным клиническим признаком синдрома Поланда является поражение большой грудной мышцы. Так, по данным N. Yiyit et al. (2015), из 113 больных аплазия всей большой грудной мышцы отмечается в 71,6 %, только ее грудино-реберной порции — в 28,3 % случаев. Помимо большой грудной мышцы также встречается поражение и других групп мышц: малой грудной, передней зубчатой, ромбовидной, трапециевидной, широчайшей мышцы спины, прямой мышцы живота [2].
T.J. David, R.M. Winter в 1985 году сообщили о семье, в которой мужчины на протяжении трех поколений имели одностороннюю аплазию большой грудной мышцы, передней зубчатой мышцы и широчайшей мышцы спины. Авторы подчеркнули, что аплазия других мышц плечевого пояса, помимо большой грудной, может иметь решающее значение для выбора тактики хирургического лечения: в частности, невозможность использования широчайшей мышцы спины для реконструкции большой грудной мышцы [13].
У большинства пациентов с синдромом Поланда грудная клетка имеет одностороннюю деформацию в связи с гипоплазией или аплазией реберного хряща (чаще II–IV или III–V ребер) [14].
У 20,3 % больных N. Yiyit et al. (2015) выявили отсутствие передней части ребер, у 7,9 % — гипоплазию ребер. Из аномалий грудной клетки наиболее часто встречалась килевидная деформация (9,7 %), значительно реже — воронкообразная (0,8 %). Наличие дефекта более одного ребра в 7 % привело к формированию грыжи легкого, у 4,4 % пациентов было выявлено парадоксальное дыхание [2]. J. Lieber et al. (2012) описали наличие на стороне поражения дефекта передней части V ребра, гипоплазию VI, конкресценцию VII–VIII ребер [15].
N. Yiyit et al. (2015) выявили сколиоз в 1,7 % наблюдений. У больных с левосторонним проявлением синдрома Поланда N. Yiyit et al. (2015) ни в одном случае не встретили полного проявления situs inversus (в 7 % случаев была выявлена только декстракардия). У более половины пациентов с декстракардией наблюдалось частичное отсутствие ребер на стороне поражения. Пороков сердца не было диагностировано ни в одном случае [2].
У подавляющего большинства пациентов наблюдалось истончение подкожно-жировой клетчатки на стороне поражения (86,7 %). Также характерными симптомами являлись дефицит волос на грудной клетке (59,2 %) и подмышечной впадине (64,6 %) на стороне поражения [2].
В большинстве случаев у пациентов с синдромом Поланда отмечались аномалии развития грудной железы и сосково-ареолярного комплекса на стороне поражения (смещение их вверх (57,5 %), гипомастия (64,6 %), гипотелия (61 %), амастия и ателия (2,6 %), полителия (0,8 %)) [2].
Оценка степени тяжести деформации грудной клетки представляет сложности ввиду полиморфности клинических проявлений синдрома Поланда.
A.E. Seyfer et al. (2010) в зависимости от характера поражения грудной клетки выделяют две формы синдрома: простую (имеется только дефект мягких тканей) и сложную (дефект мягких тканей и каркаса грудной клетки) [16].
В настоящее время в клинической практике наиболее распространена классификация Foucras с выделением трех степеней тяжести [1].
Степень 1: незначительная деформация, обусловленная гипоплазией большой грудной мышцы и умеренной гипоплазией молочной железы, приводящая к асимметрии молочных желез у женщин и едва заметной асимметрии грудной клетки у мужчин. Сосково-ареолярный комплекс меньше по размеру, чем на здоровой стороне, и расположен выше. Отсутствуют какие-либо скелетные аномалии.
Степень 2: умеренная деформация с заметной аплазией большой грудной мышцы, гипоплазией других грудных мышц (передней зубчатой, наружной косой и широчайшей мышцы спины), умеренной деформацией ребер и заметной деформацией грудной клетки как у женщин, так и у мужчин. Имеется тяжелая гипоплазия или полная аплазия тканей молочной железы, сосково-ареолярный комплекс также гипоплазирован или полностью отсутствует.
Степень 3: тяжелая деформация грудной клетки, обусловленная не только аплазией молочной железы, большой грудной мышцы, но и других мышц грудной клетки. Имеется аплазия ребер и деформация грудины.
Врожденные аномалии развития кисти характерны для полного синдрома Поланда и выявляются на той же стороне, что и поражение грудной клетки. Частота поражения кисти, по данным разных авторов, составляет 12–56 % [1, 2]. Тяжесть их проявления различна: от укорочения средних фаланг пальцев в сочетании с синдактилией до полного отсутствия пальцев кисти. В 1989 году Shamberger et al. сообщили, что 67 % исследованных ими пациентов с синдромом Поланда имели одностороннюю синдактилию, с наиболее частым поражением II–IV пальцев кисти [17, 18]. В литературе встречаются различные классификации врожденных пороков развития верхней конечности при данном синдроме, однако наиболее полной является классификация Al-Qattan (2001), включающая выделение семи типов.
Тип 1 — нормальная кисть, отмечается лишь деформация грудной клетки.
Тип 2 — деформация кисти незначительная, проявляется только при сравнении с контралатеральной стороной.
Тип 3 — классическая форма (симбрахидактилия).
Тип 4 — на кисти имеются несколько функционирующих лучей: А — лучевая косорукость с болтающимся большим пальцем или аплазия первого луча; В — адактилия II пальца; С — адактилия II–III пальцев; D — адактилия центральных лучей (расщепление кисти); Е — адактилия ульнарных пальцев.
Тип 5 — все пальцы не функционируют или отсутствуют.
Тип 6 — поперечные дефекты проксимальнее пястно-фаланговых суставов.
Тип 7 — деформация верхней конечности, сходная с фокомелией [19].
Помимо аномалий костно-мышечной системы при синдроме Поланда возможно вовлечение в патологический процесс других органов и систем. Так, например, описаны случаи поражения кроветворной системы [20], такие как лейкемия [21] и неходжкинская лимфома [22]. Jacob Ndas Legbo в 2006 году описал случай 12-летней девочки с синдромом Поланда и тяжелой тромбоцитопенией. В анамнезе у пациентки были частые кровоизлияния в область передней грудной стенки со стороны поражения. Объяснить такую избирательность поражения автору не удалось [23].
Из сопутствующей ортопедической патологии N. Yiyit et al. (2015) у 15,9 % пациентов наблюдали болезнь Шпренгеля [2].
Лечение
Тактика лечения пациентов с синдромом Поланда определяется формой патологии (полная и неполная), степенью тяжести анатомо-функциональных изменений, а также возрастом.
При этом можно выделить две основные группы хирургических вмешательств.
- Реконструктивные вмешательства на кисти (верхней конечности).
- Реконструктивные вмешательства на грудной клетке.
Реконструктивные вмешательства на кисти (верхней конечности)
У больных с полной формой синдрома Поланда хирургическое лечение начинается с восстановления функции схвата кисти и улучшения возможности самообслуживания.
Основными вариантами вмешательств на кисти являются: устранение синдактилии, восстановление оппозиции первого луча, удлинение пальцев кисти, при крайних формах недоразвития производится пересадка пальцев стопы на кисть [24, 25].
Реконструктивные вмешательства на грудной клетке
В литературе встречаются различные мнения о сроках начала лечения пациентов с синдромом Поланда. Одни авторы считают, что операции следует проводить по окончании подросткового возраста, что позволит достичь максимальной симметрии и предотвратить последующие вмешательства [2, 29], другие описывают хорошие результаты лечения детей более раннего возраста [30, 31].
Методы коррекции деформаций грудной клетки можно разделить на следующие группы:
- восстановление контуров грудной клетки (аутотрансплантация кожно-жировых и мышечных лоскутов; липографтинг; инъекции раствора полимерного полиакриламида, протезирование);
- реконструкция грудной железы (аутотрансплантация кожно-жировых и мышечных лоскутов; протезирование; липографтинг; формирование сосково-ареолярного комплекса);
- восстановление каркасной функции грудной клетки (восстановление дефектов ребер, устранение грыжи легкого, а также деформаций грудины).
На сегодняшний день в мире при реконструкции деформации грудной клетки у пациентов с синдромом Поланда отдают предпочтение пересадке широчайшей мышцы спины (в свободном и несвободном вариантах) в позицию большой грудной мышцы [3]. По мнению N.A. Popodopulos et al. (2011) широчайшая мышца спины позволяет восполнить необходимый объем мягких тканей грудной клетки, сходный с большой грудной мышцей, и в итоге дает наилучшие результаты лечения [3]. Транспозиция широчайшей мышцы спины позволяет не только восстановить контуры, но и стабилизировать грудную клетку, а также обеспечивает дополнительное покрытие протеза железы (при его использовании) и повышает его стабильность [29]. В тех случаях, когда имеется дефицит мягких тканей на передней поверхности грудной клетки, первым этапом производится экспандерная дермотензия, а далее, после создания необходимого запаса мягких тканей, выполняется пересадка лоскутов при необходимости в сочетании с установкой импланта грудной железы. При отсутствии широчайшей мышцы на стороне поражения возможна ее пересадка с контралатеральной стороны [32].
N.A. Popodopulos et al. (2011) оценили качество жизни 49 женщин с синдромом Поланда, которым выполнялись различные вмешательства на грудной клетке: 16 — транспозиция широчайшей мышцы спины, 12 — экспандерная дермотензия с последующей установкой имплантата молочной железы, 4 — перемещение TRAM-лоскута (лоскут на основе прямой мышцы живота). Шестнадцать пациентов были удовлетворены полученными результатами (13 больным была выполнена транспозиция широчайшей мышцы спины, 2 — установка имплантата молочной железы, 1 — перемещение TRAM-лоскута).
Альтернативным методом эстетической коррекции деформации грудной клетки при синдроме Поланда является использование силиконовых имплантов. Так, Fekih et al. (2010) данную методику применяли при лечении деформации II–III степеней [39, 40]. G. Soccorso et al. (2015) сообщили об опыте лечения воронкообразной деформации грудной клетки у ребенка с синдромом Поланда силиконовым имплантом, при этом авторами был получен хороший косметический результат [31]. Однако, по данным A.E. Seyfer et al. (2010), использование силиконовых имплантатов дает большой процент осложнений [16], и, кроме того, при сложных формах синдрома постановка имплантата крайне затруднительна [36].
Одним из методов косметической коррекции деформаций грудной клетки является микроинъекционная аутотрансплантация жировой ткани, или липографтинг. Это процедура, при которой жировая ткань пациента с бедер, живота, ягодиц забирается с помощью липосакции и вводится в ту область, где это необходимо. Основными преимуществами данного способа коррекции являются отсутствие реакции отторжения, малоинвазивность, минимальный период реабилитации [41].
Однако вследствие резорбции жировой ткани с целью получения удовлетворительного эстетического результата требуются многократные вмешательства [38].
Fekih et al. (2010) описали опыт выполнения липосакции с контралатеральной стороны для устранения асимметрии грудной клетки у пациентов со среднетяжелыми деформациями. В ряде случаев с целью максимальной коррекции формы и размера грудной железы требуется выполнение редукционных вмешательств с контралатеральной стороны [39].
J. He et al (2016) описали методику одномоментного восстановления соска (за счет перфорантой торакодорзальной артерии — так называемый TAP-лоскут (кожно-жировой)) и самой железы за счет мышечного лоскута на основе широчайшей мышцы спины у пациентов с синдромом Поланда. В ходе операции под мышцу был установлен тканевой экспандер, и спустя неделю начата дермотензия. Через 2 месяца был выполнен второй этап — удаление экспандера и установка имплантата необходимого размера. Данная методика была применена у 12 пациенток в возрасте от 15 до 21 года, при этом хорошие и очень хорошие результаты были отмечены в 11 случаях [42].
В тех случаях, когда у пациентов имеются тяжелые аномалии развития грудины и ребер, вызывающие функциональные или грубые косметические нарушения, первоначально выполняются реконструктивные операции на каркасе грудной клетки: торакопластика по Равич и ее модификации [29]; замещение дефектов хрящевыми аутотрансплантатами (реберный хрящ, заимствованный с противоположной стороны), костно-хрящевыми аллотрансплантатами, искусственными материалами, титановым mesh [15]; модифицированная торакопластика по Nuss [30]; костно-пластические вмешательства, включающие одномоментное выполнение остеотомии грудины и транспозиции ребер с фиксацией системой VEPTER [15]; замещение дефекта ребер по передней поверхности грудной клетки титановыми пластинами системы Matrix rib [26].
A.E. Seyfer et al. (2010), проанализировав отдаленные результаты лечения 63 больных с синдромом Поланда в сроки наблюдения от 1 до 21 года после операции, пришли к выводу, что при простых формах синдрома показана транспозиция широчайшей мышцы спины в позицию большой грудной у мужчин и дополнение данного вмешательства у женщин установкой имплантата молочной железы под перемещаемую мышцу. В случае сложных форм помимо перемещения широчайшей мышцы спины необходимо восстанавливать и каркасную функцию грудной клетки [16]. Подобного алгоритма лечения придерживаются и L. Foucras et al. (2003). Авторы указывают на целесообразность применения липографтинга как дополнительной процедуры с целью улучшения симметрии груди [1].
Заключение
Таким образом, представленный обзор литературы позволил всесторонне осветить вопросы этиопатогенеза, клиники, современных технологий реабилитации пациентов с синдромом Поланда и выявить преимущества и недостатки существующих методик. Учитывая редкую частоту встречаемости, сложность патологии и полиморфность клинических проявлений, отсутствие единого подхода к лечению, данная тема нуждается в дальнейшем изучении и публикации новых случаев.
Информация о финансировании и конфликте интересов

Малораспространенной генетической патологией, характеризующейся изменением соединительных тканей, является синдром Марфана. Людей с классическими признаками этого заболевания несложно узнать по характерной внешности. Практически все они отличаются аномально высоким ростом и астеническим телосложением, удлиненными конечностями и пальцами, чрезмерно подвижными суставами. Заболевание характеризуется разнообразными патологическими изменениями в строении скелета, сердечной мышцы и сосудов, органов зрения. Частота появления синдрома Марфана невелика и составляет единицу на 10-20 тысяч новорожденных детей, причем на этот показатель не влияют половые или расовые особенности.
Особенности и причины заболевания
Начальные признаки синдрома Марфана появляются еще в дородовом периоде развития. Они обусловлены нарушениями развития соединительных тканей, которое вызывает мутация гена, регулирующего выработку одного из основных белков – фибриллина. Из-за структурных изменений и недостаточности фибриллина ткани становятся менее плотными и упругими, плохо переносят нагрузки. Наиболее сильно из-за этого страдают суставы и связки, стенки сосудов и глазной аппарат, в котором ослабляется ткань цинновой связки.
Основной причиной синдрома Марфана является аутосомно-доминантное наследование мутации, т. е. заболевание передается от одного из родителей к ребенку. Кроме того, в некоторых случаях изменения в генной структуре появляются из-за воздействия на женщину внешних неблагоприятных факторов, в число которых входят радиация, ионизированное излучение, а также лучевая терапия, которой мать подвергалась при лечении онкозаболевания.
Медики выделяют стертую и выраженную формы заболевания. В стертой форме изменения присутствуют в одной или двух системах, причем они довольно незначительны. При выраженной форме болезни изменения присутствуют, как минимум, в трех системах, независимо от степени их выраженности, либо в одной-двух, но достаточно ярко выражены. Состояние больного может оставаться стабильным в течение многих лет, либо патология прогрессирует, охватывая новые участки тела, системы и органы.
Основные признаки патологии

Часто внешние симптомы синдрома Марфана проявляются уже в первые дни после рождения ребенка и в дальнейшем лишь усиливаются. Среди внешних признаков, по которым можно заподозрить патологию, следует отметить, в первую очередь:
- увеличенную длину конечностей и пальцев (долихостеномелию и арахнодактилию);
- недостаточный вес при повышенном физическом развитии ребенка;
- удлиненную форму черепа и вытянутое лицо;
- слабые, плохо развитые мышечные ткани, недостаток жировой клетчатки;
- аномально высокую гибкость суставов;
- неловкость и неуклюжесть движений.
Синдром Марфана у детей старше четырех лет приводит к изменению формы грудной клетки, искривлению позвоночника, развитию плоскостопия.
Среди офтальмологических симптомов наиболее часто присутствует близорукость, эктопия глазного хрусталика, изменение формы роговицы, косоглазие, гипоплазия радужки и сетчатки. Изменения часто проявляются уже в первые годы жизни и носят двусторонний характер, устойчиво прогрессируя с течением времени.
Наиболее опасными являются патологические изменения сердечно-сосудистой системы, которые при отсутствии медицинской помощи приводят больного к летальному исходу в раннем возрасте. Сюда относятся изменения сосудистых стенок, различные пороки структуры сердца и коронарных сосудов. При наиболее неблагоприятной форме заболевания у ребенка уже на первом году жизни развивается прогрессирующая сердечная недостаточность.
Кроме того, симптомы синдрома Марфана могут проявляться в работе других систем и органов. Болезнь может поражать нервные ткани, бронхи и легкие, кожные покровы, мочевыделительную и половую систему.
Как точно определить патологию у ребенка?
В настоящее время диагностика синдрома Марфана базируется на соответствии клинической картины Гентским критериям, разработанным в 1995 году и уточненным в 2010 году. Они описывают ряд признаков патологии для костно-скелетной системы, органов зрения, сердца и сосудов, а также для других систем и органов. Чтобы определить степень соответствия, врач собирает анамнез (в том числе семейный), проводит тщательный осмотр больного с проведением фенотипических тестов, назначает лабораторные анализы и инструментальные исследования, в число которых входят:

- анализ мочи на определение гликозаминогликанов;
- выявление генотипа ДНК;
- проведение ЭКГ и ЭхоЭКГ для выявления патологий сердца и сосудов;
- проведение УЗИ сердца;
- рентген грудной клетки для фиксации деформаций скелета, сердца и легких;
- компьютерная и магнитно-резонансная томография.
При необходимости могут быть назначены другие анализы и исследования.
Как лечат?
Поскольку заболевание имеет генетическую природу, и на сегодняшний день медицина не обладает инструментами для исправления генных мутаций, то лечение синдрома Марфана направлено на улучшение состояния пациента, купирование прогресса болезни и устранение клинических проявлений. Это комплексный процесс, в котором принимают участие разные специалисты, в зависимости от характера наиболее выраженных симптомов – ортопед, кардиолог, офтальмолог, терапевт, врачи других направлений.
Клинические рекомендации при синдроме Марфана включают ограничение физических нагрузок до минимально допустимого уровня, чтобы избежать развития патологий сердца и сосудов, пневмоторакса и других опасных состояний. Лечебные усилия включают:
- прием лекарственных препаратов;
- при необходимости – хирургические вмешательства для коррекции наиболее поврежденных участков сердца и системы кровообращения;
- ортопедическую коррекцию;
- санаторное лечение, физиопроцедуры, лечебную физкультуру.
При соблюдении врачебных рекомендаций прогноз практически всегда благоприятен: усилиями медиков течение заболевания существенно облегчается, они получают возможность прожить долгую жизнь без серьезных осложнений здоровья.
Часто возникающие вопросы
Можно ли предупредить развитие синдрома Марфана у ребенка?
Если хотя бы у одного из супругов в семейном анамнезе встречался синдром Марфана, пара перед зачатием ребенка должна пройти генетическое обследование. После наступления беременности проводится пренатальная диагностика: выполняется УЗИ плода и ряд биохимических исследований: материнской сыворотки, околоплодных вод, биопсия хориона, исследование клеток плаценты и пуповинной крови.
Берут ли в армию с синдромом Марфана?
Нет. Диагностированный у призывника синдром Марфана является основанием для освобождения от службы в армии.
Каковы меры профилактики при синдроме Марфана?
Пациенты в течение всей жизни находятся под врачебным наблюдением. Профилактические меры направлены на предупреждение осложнений, для чего проводится кардиохирургическая коррекция, медикаментозная терапия устраняет риск тромбирования сосудов, проводится антибактериальная терапия и т.д.
У современных подростков в результате активизации роста костной и мышечной ткани могут происходить патологические изменения в развитии опорно-двигательного аппарата. Если своевременно обратиться к врачу, то можно все отклонения нормализировать, а при отсутствии лечения могут развиваться заболевания позвоночника, одним из которых является болезнь Шейермана-Мау. В связи с этим, многих призывников интересует, берут ли в армию с Шейермана-Мау?
Особенности заболевания
Болезнь Шейермана-Мау.- это искривление позвоночника в грудном отделе, которое опасно тем, что его очень сложно диагностировать на раннем этапе, чтобы купировать наметившуюся патологию в развитии позвоночного столба.
Здоровый позвоночник имеет S-образную форму. В нем имеется 4 отклонения от вертикальной оси – 2 вперед и 2 назад, которые выполняют компрессионную функцию, равномерно распределяя нагрузку. Такая конфигурация позволяет позвоночнику действовать как пружина во время нагрузок. Норма искривления вперед и назад не должна превышать 20 и 40 градусов. При увеличении угла изгиба в грудном отделе меняется не только осанка, но и расположение самих позвонков, которые приобретают клиновидную форму, в результате чего соединяющая их хрящевая прослойка быстро истончается, и в грудном отделе начинают образовываться многочисленные позвоночные грыжи.
Позвоночник теряет свою подвижность, начинает давить на легкие, бронхи и сердце, снижая их рабочие функции. По этой причине заболевание Шейерман Мау и армия не могут совмещаться из-за того, что молодой человек теряет нужную подвижность, на фоне такого искривления позвоночника могут развиваться вторичные патологии внутренних органов. В результате искривления может произойти защемление нервных окончаний и сосудов, что может вызвать инвалидизацию и даже летальный исход.
Проконсультируйтесь
Найдите ответы на свои вопросы или обратитесь к военному адвокату за консультацией
Стадии заболевания
Болезнь Шейермана-Мау врачи делят на несколько стадий:
Скрытая стадия еще не означает патологических изменений в строении позвоночного столба, но ее сложно выявить. На ранней стадии ребенка могут мучить боли в области груди из-за того, что начинают развиваться позвоночные грыжи. На поздней стадии может потребоваться хирургическое вмешательство, так как искривление грудного отдела может пережимать важные нервы и сосуды.
Годность к военной службе
Военкоматы разработали специальные нормативы для болезни Шейерман-Мау и категории годности призывников. Само такое заболевание опорно-двигательного аппарата не является причиной отсрочки от армии, кроме тяжелых форм протекания последней стадии.
Врачи из медкомиссии признают годными к воинской службе призывников с начальными стадиями заболевания. При наличии ранней и поздней формы протекания такой патологии позвоночника в нетяжелой форме молодому человеку могут дать отсрочку от армии для прохождения лечения, после чего он может быть признан ограниченно годным для воинской службы.
При определении категории годности при наличии такого диагноза опорно-двигательного аппарата медики всегда индивидуально подходят к каждому призывнику. Чтобы получить отсрочку от военной службы, следует предоставить больше медицинских заключений, результатов диагностики и направлений на лечение от специалистов.
При наличии у призывника тяжелых осложнений, не позволяющих ему выполнять в полном объеме физическую нагрузку, предусмотренную службой в армии, его могут полностью комиссовать. Для этого требуется собрать как можно больше медицинских заключений, свидетельствующих о наличии тяжелых осложнений, вызванных заболеванием Шейермана-Мау.
Если у вас есть сомнения, можете ли рассчитывать на освобождение от армии или на службу без ограничений в связи с состоянием здоровья, то лучше проконсультируйтесь с опытным военным адвокатом, который поможет разобраться в нюансах и выработать правильную линию поведения.
Читайте также:
- Как обновить программное обеспечение на компьютере виндовс 7
- Основы православной культуры в школе для чего и как пособие для родителей и учителей
- Чем компенсировать недостаток секса
- Какие финансовые риски больше всего угрожают благополучию современных российских семей
- Тасс уполномочен заявить что партийные и государственные деятели какой год