
Паучьи пальцы как наследуется
Обновлено: 17.05.2024
Синдром Марфана (Marfan). Синонимы: арахнодактилия, гиперхондроплазия. Это одно из самых частых наследственных заболеваний соединительной ткани с множественными висцеральными аномалиями, среди которых наиболее важными являются поражения гистологических структур сердца, крупных сосудов, опорно-двигательного аппарата и глаз.
Заболевание обусловлено мутацией гена фибриллина на 15 хромосоме; имеет аутосомно-доминантный тип наследования с высокой пенетрантностью мутантного гена. В результате нарушается синтез коллагена и эластина, а накопление повышенного количества аномального (незрелого) коллагена приводит к формированию характерных фенотипических проявлений болезни.
Частота диагностированных клинических форм синдрома Марфана (СМ) в популяции составляет 1 на 10-15 тысяч человек и не зависит от географических или этнических факторов. Однако несомненно, что неустановленных случаев СМ и так называемых мягких форм болезни гораздо больше. Признаки СМ выявляются у одного из родителей больного в 60-70% наблюдений, у остальных заболевших обнаруживается новая мутация гена F 1 BN 1. Выраженная экспрессивность доминантного гена порождает обширный спектр клинических проявлении болезни – от крайне тяжелых до трудно диагностируемых стертых форм заболевания. Такое может наблюдаться в рамках одной семьи.
Полная клиническая картина СМ встречается редко, при этом наблюдается явная возрастная динамика самых постоянных симптомов болезни. В периоде новорожденности удается отметить только признаки арахнодактилии. Астеническая конституция формируется к 3-му году, а характерный внешний облик возникает к 3-4 годам. Выраженные скелетные изменения становятся очевидными лишь к 16 годам.
Больные жалуются на ноющие боли в спине, в костях. Дети с СМ начинают ходить с запозданием, у них при этом часто возникают вывихи суставов. Пациенты с типичным СМ имеют высокий рост, несоразмерно длинные конечности. Изменения трубчатых костей наиболее типичны. Пальцы рук тонкие и длинные – “паучьи пальцы”, “пальцы мадонны”.
Нередко отмечается слабость межреберных мышц, что способствует возникновению деформаций грудной клетки. Они носят своеобразный характер: западение на уровне грудины (воронкообразная грудная клетка), протрузия грудины (килевидная грудь), асимметрия грудной клетки, “крыловидные” лопатки.
Нередко встречаются высокое нёбо, волчья пасть, гиподонтия, симптом “набегания” зубов. Типичны сколиоз, кифосколиоз. Выраженные изменения грудной клетки могут явиться причиной инвалидности.
При наличии гипермобильности суставов ее степень бывает невелика (в отличие от синдрома Элерса-Данло). Пальцы рук и ног могут быть искривлены. Типичны экзостозы пяточных костей, плоскостопие, поперечное и продольное. Изредка наблюдаются подвывихи и вывихи пальцев кистей и стоп: это объясняется слабостью связочного аппарата. Помимо деформаций нижних конечностей, можно обнаружить у части больных СМ врожденный артроз бедра, анкилозы пальцев конечностей, проявления остеоартроза других суставов.
Весьма характерны трофические расстройства кожи: мраморность, цианоз пальцев конечностей, гипергидроз или сухость кожи, ломкость ногтей.
Особенно ответственными в диагностике СМ являются поражения сердца и сосудов: от их выраженности зависит прогноз для жизни этих больных. Аномалии сердечно-сосудистой системы отмечаются у 50-90% больных СМ. Изменения эластического каркаса всех сосудов носят генерализованный характер. Но наиболее значимыми являются изменения стенки аорты: чаще всего поражается начальная часть ее, т.е. клапанное кольцо и синусы Вальсальвы. В итоге развивается аортальная недостаточность, кардиомегалия, левожелудочковая недостаточность. При прогрессировании изменений возникает аневризма аорты. По этой причине больные, в особенности занимающиеся физической работой, спортом, могут внезапно погибнуть от разрыва или расслоения аорты. Этому также способствует артериальная гипертензия. До 60% больных СМ погибают от разрыва аорты в молодом возрасте.
Опасны и другие осложнения СМ (инфекционный эндокардит, аневризма межпредсердной перегородки). Пролапс митрального клапана – тоже типичное проявление СМ, так как при этом клапаны сердца топкие, легкорастяжимые. Сердце может приобрести вид “капельного”.
Больные СМ с поражением ССС предъявляют жалобы на одышку при физической нагрузке, боли в сердце (длительные, до нескольких часов), перебои в сердце.
При неглубоком обследовании у них ошибочно диагностируют врожденные или ревматические пороки сердца, обычно – митральную недостаточность, сифилитический аортит. Симптомы со стороны ССС вариабельны: от функционального шума, пресистолического щелчка, лабильности пульса и АД, до грубых систоло-диастолических шумов над аортой и легочной артерией. Но отмечены варианты поражения аорты, когда, без прогрессирования аневризмы или порока сердца, эти изменения остаются незамеченными в течение всей жизни больных.
Типичными могут оказаться и изменения со стороны органа зрения: вывих (подвывих) хрусталика, что приводит к миопии, спазмам аккомодации, реже встречаются афакия, вторичная глаукома, отслойка сетчатки, миопический астигматизм, дегенерация желтого пятна, так называемые синие склеры и др. Глазные проявления обычно выявляются после 18 лет, за исключением рано обнаруживаемого подвывиха хрусталика.
В рамках СМ обнаруживаются дизгистогенетические аномалии легких: буллезная эмфизема легких, спонтанный пневмоторакс, выраженный фиброз, уплотнения корней легких.
Из важных врожденных аномалий ЦНС отмечаются расширение твердой мозговой оболочки, гидроцефалия, расширение большой цистерны мозга, анизокория. С детских лет заметно нарушение внимания, отставание в учебе, инфантилизм, у девочек – нарушения менструаций. Отмечаются также асимметрия рефлексов, пирамидные симптомы, нистагм.
При поражении органов ЖКТ типичны длительные парезы кишечника. Если СМ сопровождается аномалиями мягких тканей, то заметны гипоплазия мускулатуры и жировой ткани, перерастяжимость сухожилий и суставов.
Важнейшими, не могущими остаться незамеченными, являются аномалии внешнего облика больных СМ: большой нос, выраженный подбородок, что придает лицу “птичье” выражение, дисплазия ушных мочек, антимонголоидный разрез глаз, старческий вид – у детей.
Важно отметить, что полного набора даже кардинальных признаков СМ у конкретных больных практически не встречается, а прогрессирование болезни и трансформация ведущих симптомов СМ могут продолжаться на протяжении всей жизни больного. Дифференциальный диагноз СМ проводится со сходными аномалиями соединительной ткани – С. Элерса-Данло, MASS -синдромом, марфаноподобным семейным фенотипом и др.
Диагностика СМ практически зиждется на выявлении клинических проявлений болезни. Подтверждают диагноз анализ родословной, осмотр членов семьи клиническим генетиком.
Диагностика.
Подтверждают диагноз СМ повышенное содержание и выведение оксипролина, но при стабильной форме заболевания возможно полное отсутствие изменений концентрации оксипролина, либо это является доказательством наличия подобного, но другого заболевания. Вдвое увеличена почечная экскреция гликозоами-ногликанов.
Характер поражения ССС и тяжесть его оцениваются по ЭХО-кардиографическому исследованию (аневризмы различных отделов аорты, увеличение полостей предсердий и левого желудочка) и допплерографическим данным. Проводятся при необходимости УЗИ органов брюшной полости и КТ исследование.
Лечение.
У детей с незавершенным остеогенезом проводится так называемая коллагеннормализующая терапия (бета-адреноблокаторы, рибофлавин, витамины С и пиридоксин). Взрослым проводится так называемая ситуционная терапия бета-блокаторами, препаратами калия, сердечными гликозидами, мочегонными. Санируются очаги инфекции.


Типичным визуальным признаком недуга являются тонкие и длинные пальцы, несколько напоминающие конечности паукообразных. Заболевание сопровождается нарушением двигательных возможностей суставов пальцев рук, повышенной их гибкостью, эластичностью.
Как правило, недуг проявляется при наследственных болезнях, мутациях гена, генетических заболеваниях. Однако вполне допустим и врожденный дефект развития пальцев, выраженный в их чрезмерной длине и гибкости, который является анатомической индивидуальной особенностью и не сопровождается развитием патологических состояний.
Что это такое
Преимущественно симптом паучьих пальцев проявляется на фоне совокупных заболеваний, к числу которых относят:
- Синдром Марфана. При данном синдроме наблюдается также нарушение функционирования различных систем и органов, изменение сердечно сосудистой системы, в частности – сердечных клапанов.
- Гомоцистинурия. Системное заболевание, отнесенное к числу генетических патологий.
Для выявления истинной причины паучьих пальцев и определения необходимости назначения лечения требуется проведение полного комплексного обследования пациента.
Видео
Причины
В настоящее время существует масса теорий относительно причин развития арахнодактилии. К числу наиболее вероятных причин относят нарушение обмена незаменимой аминокислоты метионина, вызванного полным отсутствием в организме или недостатком ряда ферментов, к числу которых, например, относится белок фибриллина.
Симптомы
Синдром паучьих пальцев сопровождается проявлением ряда характерных симптомов, основным среди которых является патологическое удлинение отдельных частей тела. Особенно подвержены поражениям такого рода кисти, пальцы и стопы. Зачастую в полной мере клиническая картина проявляется только после достижения ребенком возраста трех – четырех лет и сопровождается следующими признаками:
- Расположение линии талии значительно выше уровня принятых анатомических норм.
- Грудная клетка является аномально узкой, наблюдается воронкообразная впадина.
- Нарушены пропорции формы черепа – костные ткани вытянуты и более узкие.
- Деформация тазовых костей.
- Возможным проявлением является нарушения структуры и функционирования мышечных тканей, связочного аппарата.
- Длинные, несоразмерно тонкие конечности.
Следует отметить, что главный признак арахнодактилии – тонкие, деформированные в области сочленения, длинные пальцы. При этом длина фаланг не является идентичной с размером сухожилий, что влечет нарушение функционирования кистей, а также их искривление.
Диагностика
Синдром паучьих пальцев ярко выражен визуально, однако для постановки более точного диагноза, а также выяснения причин развития недуга требуется тщательное обследование не только пациента, но также его близких родственников. Основными методами диагностики арахнодактилии являются:
- Общий и физикальный осмотр на предмет выявления отклонений пропорций тела от норм анатомического строения.
- Обследования отдельных систем и органов, включающие проведение ЭКГ, осмотров офтальмолога, рентгенографии, компьютерной томографии.
- Сбор анамнеза, включающий обследование родственников.
При необходимости врач может предписать пациенту прохождение дополнительных вариантов обследования, необходимых для получения более полной картины.
Лечение
Синдроме марфана или арахнодактилия является результатом генетических мутаций, в связи с чем полностью устранить проявления недуга невозможно. Однако вполне допустимым является применение комплексных мероприятий, которые помогут существенно улучшить общее самочувствие пациента, а также предупредить развитие тяжелых осложнений, которые могут стать причиной нетрудоспособности больного.
Диета
Больным с синдромом паучьих пальцев не требуется соблюдать диету, как таковую. Питание должно быть насыщенным энергетически, содержать достаточное количество витаминов, полезных веществ. Однако в индивидуальных случаях врач может предписать больному соблюдать диету, лечебное действие которой направлено на стимуляцию кровотока, обменных процессов.
Кроме того, при некоторых вариантах синдрома паучьих пальцев необходимо снижение в рационе больного количества животного белка.

Один из побочных результатов арахнодактилии – низкий мышечный тонус, слабость, нарушение структуры связочного аппарата. Для нормализации состояния пациента, а также повышения работоспособности рекомендуется регулярно выполнять простые упражнения лечебного гимнастического комплекса, которые помогут улучшить общее самочувствие, повысить тонус мышечных тканей, укрепить их. Лечебную физкультуру называют необходимым элементом комплексного лечения детей раннего возраста. При условии регулярного проведения сеансов существенно снижается вероятность развития осложнений, деформаций, различного рода патологий.
Массаж
Регулярное проведение сеансов массажа при паучьих пальцах поможет существенно улучшить общее самочувствие пациента, повысить тонус мышечных тканей, а также укрепить их, снять болезненные проявления, если таковые имеются. Важно также отметить, что регулярное выполнение массаж специалистом по отношению к пациенту, возраст которого составляет менее трех – пяти лет, позволит существенно снизить вероятность появление деформаций или диспропорций тела.
Народные средства
Применение народных средств для устранения синдрома паучьих пальцев не обладает эффективностью ввиду специфики недуга. Однако применение некоторых рецептов поможет несколько улучшить состояние пациента. К их числу можно отнести такие, например, как:
-
При определенных нарушениях больным предписывается регулярный прием аскорбиновой кислоты. Для увеличения в организме количества данного вещества рекомендуется регулярно употреблять компот, приготовленный из плодов шиповника.
Приготовить его более чем просто – достаточно заварить ложку плодов кипятком, после чего проварить на протяжении нескольких минут, остудить.

Специфика синдрома паучьих пальцев такова, что привести к негативным последствиям, ухудшению состояния пациента могут даже незначительные факторы. Для предупреждения возможных осложнений важно проконсультироваться со специалистом перед использованием любого домашнего средства.
Операция
Оперативные вмешательства на фоне паучьих пальцев являются необходимой мерой только в том случае, если заболевание сопровождается выраженными нарушениями или отклонениями, которые существенно снижают качество жизни пациента или являются причиной утраты трудоспособности. Преимущественно назначают следующие варианты хирургических вмешательств:
Немалое количество больных, страдающих арахнодактилией, проявляет также жалобы на существенное ухудшение качества зрения. В качестве дополнительных мероприятий, направленных на общее улучшение самочувствия больного, может быть проведена операция по восстановлению или улучшению зрения.
Профилактика
Как уже было отмечено выше, симптом паучьих пальцев является отдельным или входящим в комплекс патологий нарушением, основной причиной возникновения которого является генетическая мутация. Предупредить или исключить вероятность рождения ребенка с подобным заболеванием практически невозможно.
Основные меры профилактики заключаются в постановке диагноза в раннем детском возрасте и проведении соответствующего симптоматического лечения. Такая мера поможет существенно снизить вероятность развития тяжелых осложнений. При своевременном принятии мер больной с синдромом паучьих пальцев вполне может вести обычный образ жизни.
Если вы нашли ошибку, пожалуйста, выделите фрагмент текста и нажмите Ctrl+Enter. Мы обязательно её исправим, а Вам будет + к карме

Синдром Марфана — наследственное заболевание, которое проявляется системным поражением соединительной ткани в организме человека. В результате болезни происходят нарушения строения скелета и кожи, работы глаз, сердечно-сосудистой, дыхательной и других систем организма. Эту генетическую мутацию нельзя предотвратить или вылечить, но правильно подобранное лечение способно продлить пациентам жизнь и предупредить развитие опасных осложнений.
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
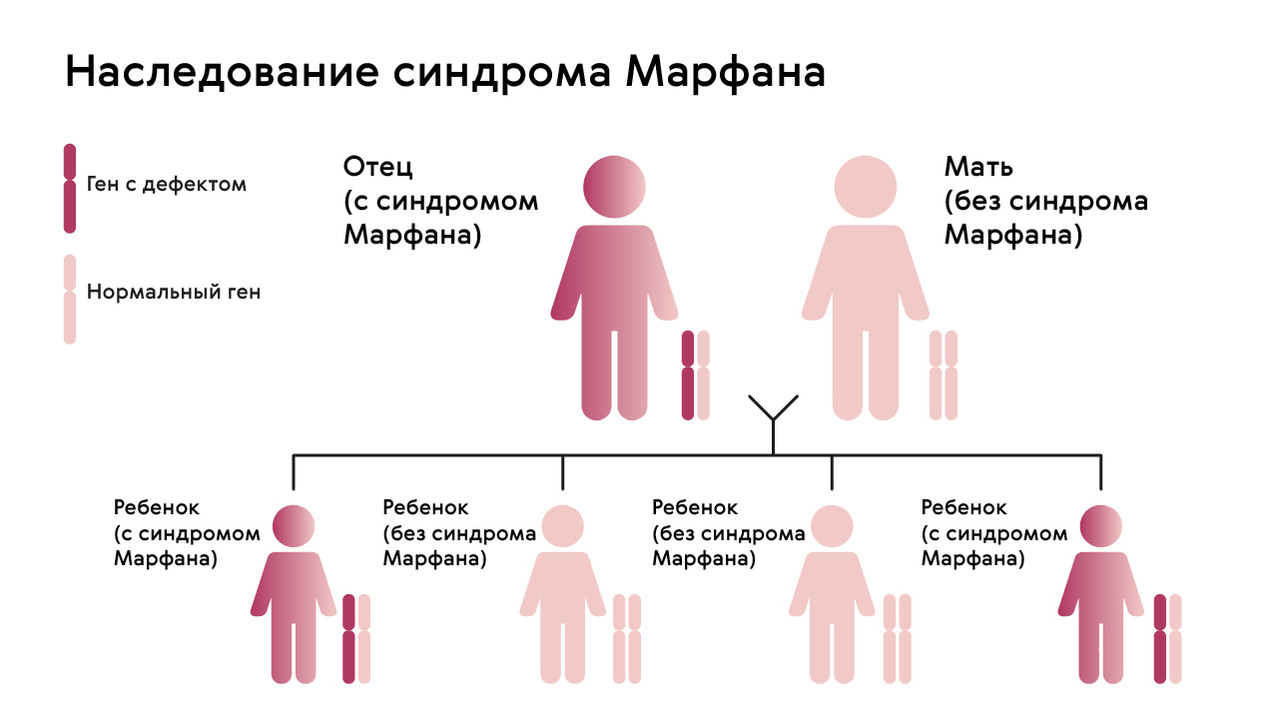
Рисунок. 1. Схема наследования синдрома Марфана. Источник: МедПортал
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на 5–10 тысяч.
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25–50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
Симптомы синдрома Марфана
Проявления генетического дефекта могут быть выражены в разной степени: от легкого изменения строения соединительной ткани до тяжелых нарушений жизненно важных функций организма. Иными словами, внешние признаки аномалии у разных пациентов могут значительно отличаться, несмотря на одинаковый генетический дефект.
Классической триадой синдрома Марфана считаются: скелетные нарушения, смещение хрусталика и расслоение аорты (рис. 2). Также системное поражение соединительной ткани у пациентов становится причиной развития нарушений работы практически всех органов и систем организма.
Костно-мышечная система
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести случая и особенностей организма пациента.
Проверить наличие симптома можно с помощью теста большого пальца кисти — у пациентов с арахнодактилией часть большого пальца (дистальная фаланга) выступает за край сжатой в кулак ладони (рис. 4).
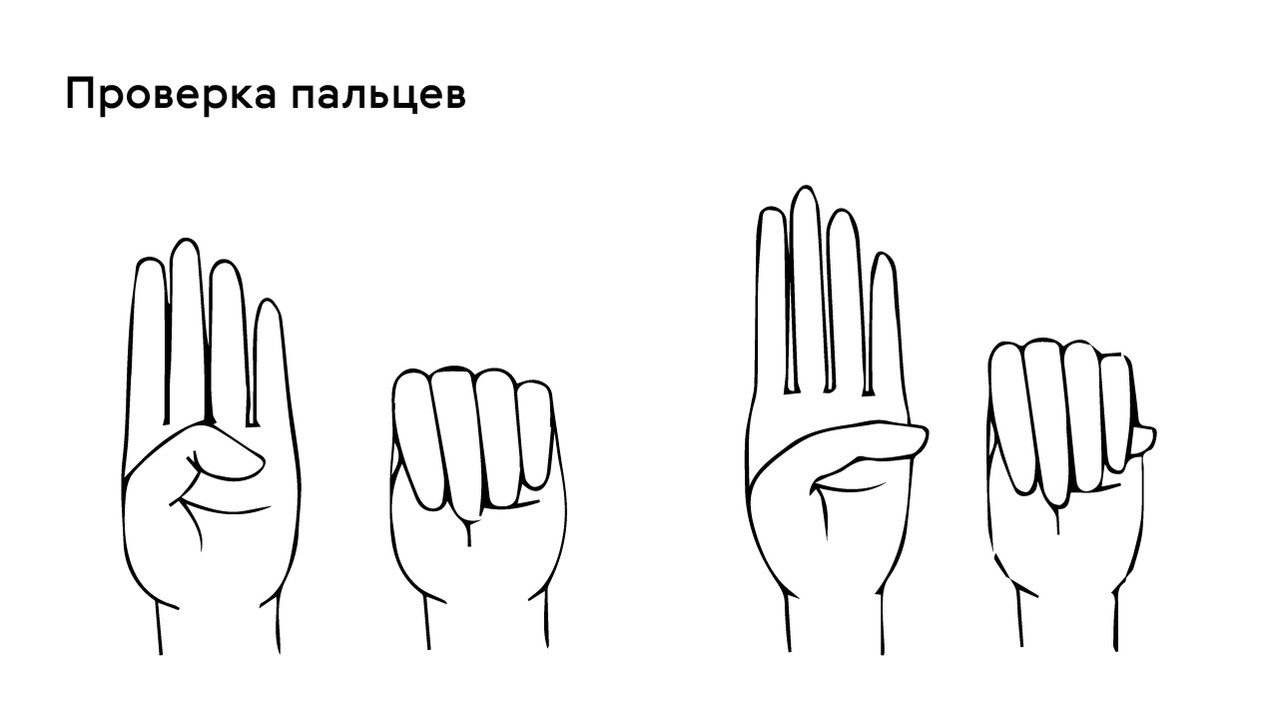
Рисунок 4. Проверка на арахнодактилию. Источник
Лицо людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
Также для таких пациентов характерны деформации грудной клетки, которые могут быть в двух вариантах: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь, рис. 5).
Кроме того, пациенты с FBN1 мутацией часто страдают от:
- плоскостопия;
- повышенной подвижности всех суставов;
- слабости связочного аппарата.
У пациентов с синдромом Марфана часто плохо развиты мышечные структуры и практически нет подкожно-жирового слоя. Движения пациентов с этой патологией неловкие, они часто получают различные травмы.
Зрение
Дефекты гена FBN1 определяют склонность к патологиям зрительной системы. Чаще всего повреждения глаз у пациентов с синдромом Марфана включают в себя:
- выраженную близорукость;
- подвывих или изменение положения хрусталика;
- высокий риск внезапной отслойки сетчатки глаза.
Кроме того, у таких пациентов гораздо раньше может развиться катаракта или глаукома: те патологии органа зрения, которые считаются возрастными у здоровых людей.
Органы дыхания
Желудочно-кишечный тракт
Процессы пищеварения у людей с FBN1 мутацией меняются: нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз.
Почечный аппарат
У пациентов с синдромом Марфана чаще находят аномалии почек: опущение органов, расширение почечных лоханок, патологическую подвижность почек.
Нервная система и психическая сфера
Хотя в большинстве случаев у пациентов с синдромом Марфана не происходит нарушений работы мозговых структур, некоторые патологические изменения нервной системы могут присутствовать. Например, расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. Для таких пациентов характерно развитие синдрома хронической усталости — астения, склонность к депрессии. Интеллектуальная деятельность в большинстве случаев не нарушена, даже наоборот: среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Сердечно-сосудистая система
Кардиологи выявляют нарушения ритма сердца у людей с синдромом Марфана. У пациентов с этой патологией часто нарушается структура аортального клапана — соединительнотканной перегородки, которая предупреждает обратный ток крови из аорты в сердце. Это приводит к развитию порока сердца — аортальной недостаточности. Также могут развиваться другие пороки сердца, например, пролапс или недостаточность митрального клапана, а на пораженных участках часто развивается инфекционно-воспалительный процесс — бактериальный эндокардит.
Самую большую опасность представляют патологические изменения в главном сосуде организма — аорте. У 65–100% людей с синдромом Марфана есть большой риск поражения луковицы (наиболее близкая к сердцу часть аорты) и восходящей дуги этой артерии — тех частей, которые непосредственно выходят из сердца. Поскольку внутренний слой стенки сосудов также содержит волокна соединительной ткани, они склонны к износу, а давление крови в аорте выше, чем в других участках сосудистого русла. Это приводит к тому, что сосуд постепенно расширяется, и может произойти патологическое скопление крови между сосудистыми стенками с формированием мешковидного выпячивания (аневризмы) или спонтанный разрыв артерии.
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка. При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).
Диагностика синдрома Марфана
Диагностика генетической аномалии включает в себя комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7–18 лет — это измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку в 10 см.
Генетическое обследование включает в себя выявление генотипа ДНК — идентификацию мутаций в гене FBN1. При возможности назначают специфические лабораторные тесты — определение выведения с мочой метаболитов соединительной ткани, таких как оксипролин и гликозаминогликаны.
Чтобы подтвердить нарушения развития соединительной ткани и оценить степень выраженности мутации гена FBN1, пациентам с подозрением на синдром Марфана назначают:
- ЭКГ;
- УЗИ сердца;
- КТ-ангиографию аорты и других сосудов;
- КТ грудной и брюшной полостей;
- МРТ позвоночника и головного мозга;
- специфические обследования на осмотре у офтальмолога;
- биопсию кожи.
Для окончательного определения диагноза используют общепринятые Гентские критерии 2010 года, согласно которым диагноз устанавливают в случаях:
- подтвержденной мутации гена FBN1 и расширения корня аорты или эктопией хрусталика;
- подтвержденного расширения корня аорты в сочетании с эктопией хрусталика;
- подтвержденной эктопии хрусталика в сочетании с любыми признаками системного поражения соединительной ткани.
Лечение синдрома Марфана
К сожалению, на сегодняшний день лекарственные методы терапии этой генетической патологии еще не разработаны. Однако пациентам с синдромом Марфана важно соблюдать все назначения врачей, чтобы устранить симптомы патологии и замедлить темпы ее развития.
Лечение зависит от клинических проявлений болезни:
- при аневризме аорты назначают препараты, которые снижают частоту и силу сердечных сокращений, снимая избыточную нагрузку на сосуды;
- пациентам с синдромом Марфана часто назначают антигипертензивные препараты для снижения артериального давления;
- хондроитин и глюкозамин относятся к естественным компонентам соединительной ткани — их прием улучшает структуру хрящей и предупреждает патологии суставов;
- для стимуляции образования коллагена выписывают специальные БАДы — L-карнитин, витамины из групп С, D, Е, В, а также кальций, цинк и другие пищевые добавки.
Пациентам противопоказаны физические нагрузки, постоянная активность, травмоопасные игры. Рацион питания людей с синдромом Марфана должен быть насыщен белками, полезными жирными кислотами, микро- и макроэлементами. Для поддержки структур скелета пациентам с мутацией в гене FBN1 показано ношение корсетов, укрепление мышц с помощью ЛФК и оздоровительного массажа.
В некоторых случаях может помочь только хирургическое лечение — операции по замене части аорты, клапанов, исправлению костных патологий или коррекции патологий глаза, которые существенно снижают риски опасных осложнений.
Прогноз
Современные методы исследования в медицине позволяют выявлять заболевание у детей в раннем возрасте. Это помогает повысить качество жизни таких пациентов и предупредить раннюю смертность. Продолжительность жизни людей с синдромом Марфана при бережном отношении к своему здоровью достигает 70 лет. Прогноз болезни во многом зависит от выраженности сердечно-сосудистых патологий, поскольку выживание пациентов с этой генетической аномалией определяет состояние аорты и риск ее спонтанного разрыва. Такие люди требуют постоянного наблюдения у врачей различных специальностей для своевременной коррекции проявлений синдрома.
Заключение
Конечно, жизнь с этой генетической мутацией становится сложнее, но при правильном подходе к собственному здоровью и своевременному обследованию у врачей пациентам с синдромом Марфана удается компенсировать все проявления заболевания и не допустить развития фатальных осложнений.
Активисты с синдромом Марфана создают тематические сообщества по всему миру: мощная поддержка людей с такой же генетической аномалией позволяет пациентам не чувствовать себя одинокими.
В числе серьезных генетических заболеваний находится синдром Марфана, которым страдает один человек из 10-20 тысяч. Им болели такие всемирно известные личности, как Авраам Линкольн, Никколо Паганини, Корней Чуковский и Ганс Христиан Андерсен.
У больного этим синдромом присутствуют различные симптомы: арахнодактилия (удлиненные кисти, паукообразные пальцы рук и ног), множественные костно-суставные аномалии, деформация грудной клетки, нарушения сердечно-сосудистой системы и т.д. В этой статье речь пойдет об одном из них — арахнодактилии, ее проявлениях, методах диагностики и способах лечения.
Что такое арахнодактилия
Причины появления
Достоверные и точные причины появления этой патологии у человека до сих пор неизвестны. Большая часть исследователей придерживается той версии, что заболевание связано с нарушением строения коллагеновых волокон и на 75% передается наследственным путем.
Остальные 25% связывают с различными генетическими мутациями, которые могут произойти спонтанно в период зачатия как в яйцеклетке, так и в сперматозоиде.
Формы проявления заболевания, общие симптомы и признаки
- Синдром Марфана. Основной вид патологии, передающийся по наследству по аутосомно-доминантному признаку. Сопровождается ярко выраженной деформацией костно-мышечной системы и осложнениями на сердечно-сосудистой системе.
- Гомоцистинурия. Генетическое заболевание, которое передается по аутосомно-рецессивному типу. Редкая патология, встречается примерно у одного человека на 200-300 тысяч.
- Непредвиденные генные мутации, вызывающие патологии различного характера.
Внешние симптомы больного арахнодактилией:
- длинные и тонкие конечности и пальцы,
- выраженный сколиоз, кифосколиоз,
- воронкообразная, суженная, удлиненная грудная клетка,
- талия, расположенная выше нормы,
- аномально высоко сформированная надколенная чашечка,
- деформированные кости таза,
- узкий удлиненный череп,
- неразвитая мышечная система, выраженная худоба,
- широко расставленные глаза,
- высокий свод неба,
- неправильное развитие глаз и другие симптомы.
Основные признаки болезни:
- многочисленные болезни клапанов сердца и аорты,
- гипоплазия легких,
- офтальмологические заболевания (близорукость, косоглазие, катаракта, подвывих хрусталика и др.),
- поражение артерий,
- остеопороз,
- разболтанность суставов,
- припадки,
- могут присутствовать отклонения в психике.
Интересно! Характерный для арахнодактилии выброс адреналина способствует развитию выдающихся умственных способностей и одаренности. Некоторые больные могут отличаться высокой силой духа и сильными волевыми качествами.
Методы диагностики
Внешние проявления заболевания легко диагностируются при помощи визуального осмотра пациента, замера длины конечностей и гибкости суставов. При обнаружении первых признаков назначается комплексное обследование для выявления сопутствующих заболеваний, патологических изменений, нарушений работы внутренних органов.
Для этого применяют следующие исследования:
- рентген позвоночника, груди и таза,
- МРТ, КТ костей и суставов,
- сцинтиграфия.
Для выявления нарушений работы сердечно-сосудистой системы назначают:
- УЗИ сердца и сосудов,
- томографию артерий и аорты,
- анализы крови.
При подозрении на гомоцистинурию проводят:
- МРТ головного мозга,
- КТ черепа и позвоночника,
- электроэнцефалографию,
- реоэнцефалографию.
Также необходимо обследовать органы зрения у офтальмолога, для чего проводят различные оптические и УЗИ-исследования.
Лечение
Лечение для каждого больного арахнодактилией подбирается строго индивидуально. Это связано с тем, что наличие симптомов или патологий проявляется по-разному. План лечебных и профилактических мероприятий подбирается в зависимости от особенностей протекания болезни. Как правило, основой становятся ортопедические мероприятия, направленные на коррекцию всех имеющихся деформаций.
Полностью устранить болезнь на сегодняшний день нельзя, но есть возможность значительно облегчить состояние больного.
В настоящее время разработан метод коллагенонормализующей терапии, благодаря которому корректируется метаболизм компонентов соединительной ткани, усиливается регенерация хрящей при хирургическом вмешательстве.
Если у человека проявляются заболевания сердечно-сосудистой системы или глаз, то в соответствии с показаниями назначается необходимая терапия по устранению или облегчению симптомов.
При наличии патологий грудной клетки проводится торакопластика – операция по достижению нормального положения грудной клетки и уменьшению ее размеров.
Медикаменты
Вне зависимости от причины возникновения заболевания пациенты находятся на постоянном диспансерном наблюдении, посещают ЛФК и массаж.
Важно! ЛФК при арахнодактилии можно применять только при невыраженных нарушениях опорно-двигательного аппарата и исключительно под наблюдением специалиста.
Массаж

Массажные процедуры при арахнодактилии назначаются индивидуально в зависимости от особенностей и патологий пациента, проводятся в комплексе и направлены на лечение и облегчение симптомов патологии.
Массаж выполняется только специально обученными медицинскими работниками, имеющими соответствующую квалификацию.
Заключение
Арахнодактилия — тяжелое и непредсказуемое заболевание, причины появления которого до сих пор не изучены до конца. Чтобы жить с такой патологией, понадобится пройти немало испытаний, проявить силу духа и терпение.
Помимо описанных в статье методов лечения, больному необходимо в качестве профилактики и повышения жизненного тонуса нормализовать сон и обязательно соблюдать все рекомендации врача по питанию и физической активности.
Читайте также: