
Куриная слепота наследуется как рецессивный признак
Обновлено: 28.06.2024
Появление на свет ребенка с врожденными дефектами развития всегда ошеломляет семью; эта тема - одна из самых тяжелых в акушерстве. Супруги в первый момент испытывают ни с чем не сравнимый психологический шок, который затем переходит в чувство вины, им кажется, что у них уже никогда не будет здорового ребенка.
Следует сразу сказать, что ребенок с врожденными пороками может появиться на свет абсолютно в любой семье - молодой, здоровой, без вредных привычек, с нормально протекающей беременностью. По данным многолетней статистики, во всем мире около 5% детей рождается с врожденными заболеваниями.
Врожденные пороки развития плода можно разделить на две большие группы - наследственно обусловленные (то есть заложенные в генах и хромосомах, передающиеся по наследству) и собственно врожденные (приобретенные в ходе внутриутробного развития). Такое деление довольно условно, так как большинство дефектов развития вызываются сочетанием наследственной предрасположенности и неблагоприятного внешнего воздействия, представляя собой мультифакториальные аномалии.
Проблема врожденных пороков развития плода очень многообразна, изучением этого вопроса занимаются различные специалисты - генетики, неонатологи, эмбриологи, специалисты по дородовой (пренатальной) диагностике. Разобраться в причинах всегда бывает непросто.
Наследственно обусловленные заболевания
В основе наследственно обусловленных заболеваний лежат мутации. Благодаря современным леденящим кровь триллерам, слово это вызывает сейчас у многих почти суеверный ужас. На самом деле латинское слово mutatio означает "изменение" - не более того. Мутация - это изменение наследственных свойств организма в результате перестроек в структурах, ответственных за хранение и передачу генетической информации. Заболевания, связанные с патологическими изменениями в хромосомах, обычно так и называют хромосомными заболеваниями. Под собственно наследственными заболеваниями понимают нарушения, обусловленные генными мутациями.
В ядрах соматических (неполовых) клеток содержится по 23 пары хромосом, из которых одна пара - половые хромосомы. У женщин эта пара состоит из двух одинаковых хромосом, условно называемых Х-хромосомами, у мужчин эти хромосомы разные - Х-хромосома и Y-хромосома. Неполовые хромосомы называются аутосомами.
В половых клетках хромосом в два раза меньше - не 23 пары, а 23 штуки.
При оплодотворении ядра яйцеклетки и сперматозоида сливаются, и будущий человечек получает полный набор хромосом, наследуя таким образом и материнские, и отцовские признаки.
Хромосомы состоят из генов. За каждый признак в организме отвечает пара генов - "мамин" и "папин". (Исключение составляет XY-пара половых хромосом у мужчин: не все гены Х-хромосомы имеют "напарников" в Y-хромосоме.) В каждой паре один ген доминирует (доминантный ген), т.е. проявляется обусловленный им вариант признака, другой - "уступает" (рецессивный ген). При неблагоприятном стечении обстоятельств оба гена в паре или один из них могут оказаться носителями патологического признака. В первом случае их "владелец", без сомнения, болен. Если же мы имеем дело лишь с одним "больным" геном, возможны два варианта: (1) за заболевание "отвечает" доминантный ген - тогда его носитель болен; (2) носитель патологического признака - рецессивный ген - тогда человек здоров (точнее, как говорят врачи, фенотипически здоров, т.е. при наличии "больного" гена в генотипе, отсутствуют какие-либо проявления болезни).
Аутосомно-доминантный тип наследования
Носитель патологического признака - доминантный ген, содержащийся в аутосоме (неполовой хромосоме). При этом типе наследования невозможно рождение больного ребенка у здоровых родителей - хотя бы один из родителей страдает от того же заболевания. При этом мальчики и девочки в равной степени подвержены заболеванию. Такие дефекты развития, как правило, бывают негрубыми и после успешной коррекции не препятствуют нормальной жизни.
Аутосомно-рецессивный тип наследования
Носитель патологического признака - рецессивный ген, содержащийся в аутосоме. При аутосомно-рецессивном механизме наследования ситуация выглядит парадоксально - у здоровых родителей вдруг появляется на свет ребенок с дефектами развития, порой тяжелейшими и даже несовместимыми с жизнью. Причина - носительство обоими супругами в скрытом состоянии мутантных рецессивных генов. При этом рождение больного ребенка не обязательно означает, что все следующие дети будут страдать тем же заболеванием. Так же, как и в аутосомно-доминантном типе, мальчики и девочки в равной степени подвержены заболеванию.
Сцепленное с полом рецессивное наследование
Пороки развития, сцепленные с полом, в основном обусловлены рецессивными мутациями в женской половой хромосоме (этот тип наследования называют еще Х-хромосомным). Такой признак всегда передается через мать - носительницу рецессивного "больного" гена (т.е. сама женщина здорова). Практически все пораженные - мужчины (у пораженного гена Х-хромосомы в Y-хромосоме отсутствует "партнер", который мог бы доминировать над ним). Больной мужчина никогда не передает заболевания своим сыновьям (ведь они получают от него "здоровую" Y-, а не мутантную Х-хромосому), однако все его дочери будут носительницами "рокового" гена.
Мы схематически описали типы наследования, чтобы дать читателю общее представление о сути этих механизмов. На самом деле все гораздо сложнее - куда менее однозначно и определенно.
В приведенной ниже таблице перечислены в качестве примера лишь некоторые из совместимых с жизнью наследственных аномалий
Механизм наследования
Лечебно-реабилитационные меры
Аутосомно-рецессивное наследование - возможно рождение ребенка-альбиноса от здоровых родителей. Частота в популяции 1:20 000
Отсутствие нормальной пигментации кожи, волос, радужной оболочки глаза
Эта наследственная аномалия не считается заболеванием в полном смысле этого слова и лечению не подлежит
Сцепленное с полом рецессивное наследование. Болеют главным образом мужчины. Передается от матери сыновьям
Заболевание обусловлено дефицитом некоторых факторов свертывания крови. Проявляется кровоточивостью
Лечение при кровотечении - переливание крови, плазмы; кровоостанавливающие средства общего действия; антигемофильный глобулин; профилактика травм и кровотечений
Сцепленное с полом рецессивное наследование. Наблюдается преимущественно у мужчин. Передается от матери сыновьям
Частичная цветовая слепота. Распространяется чаще всего на красный и зеленый цвета
Расстройство цветового зрения выявляют при помощи специальных таблиц или спектральных приборов. Дальтонизм лечению не подлежит
Хромосомная аномалия: у матери при созревании яйцеклетки под влиянием пока не выясненных причин в 21-й паре хромосом образуется 3 хромосомы вместо 2-х. Частота в популяции - 1:700
Одна из форм врожденного слабоумия. Степень психического недоразвития значительно колеблется. Больные в основном ласковы, добродушны, приветливы
Лечебная педагогика, основанная на склонности больных к подражательности. Обучение во вспомогательных школах, трудотерапия
Аутосомно-доминантное наследование, передается детям от родителей с врожденной формой заболевания
Опущение верхнего века вследствие недоразвития мышцы, поднимающей его
Врожденные мультифакториальные пороки развития
- Ионизирующее излучение (рентгеновские лучи, воздействие радиоактивных изотопов). Кроме прямого действия на генетический аппарат, ионизирующее излучение обладает токсическим эффектом и является причиной многих врожденных аномалий
- Тератогенные инфекции, т.е. инфекционные заболевания, передающиеся от матери плоду
- Медикаменты. Нет лекарств, которые могут быть безоговорочно признаны полностью безопасными, особенно на ранних стадиях беременности. Во время беременности лучше воздержаться от приема лекарств любого типа - за исключением, конечно, случаев, когда это необходимо для спасения жизни или ликвидации серьезной угрозы для здоровья матери или плода
- Алкоголь. Употребление алкоголя приводит к разнообразным врожденным нарушениям, выраженность которых зависит от количества употребляемого алкоголя - особенно на ранних стадиях беременности. Фетальный (т.е. поражающий плод) алкогольный синдром - тяжелейшее врожденное заболевание, порой несовместимое с жизнью.
- Никотин. Курение сигарет во время беременности приводит к отставанию ребенка в физическом развитии
- Воздействие токсических химических веществ
Зачастую, однако, в развитии врожденных пороков играет роль такой фактор, как наследственная предрасположенность: известно, что если у родителей или ближайших родственников наблюдались врожденные пороки развития, то риск родить ребенка со сходными дефектами повышается, то есть речь идет "о семейном накоплении" аномалий развития. Так, у женщины с врожденным пороком сердца шансы родить ребенка с дефектом развития сердечно-сосудистой системы несколько выше, чем у всех остальных женщин. Поэтому принято говорить не столько о просто врожденных, сколько о врожденных мультифакториальных пороках развития. Тем не менее, на большом статистическом материале показано, что повторный риск рождения ребенка с врожденным пороком развития невелик - в среднем 2-4%.
Приведем несколько примеров совместимых с жизнью врожденных мультифакториальных пороков развития
Дефект развития
Выхождение внутренних органов или глубоких тканей из полостей, обычно занимаемых ими, под кожу или в межмышечную клетчатку без нарушения целости покровов
Массаж, в случае его неэффективности - хирургическое лечение
Врожденный вывих и врожденная дисплазия тазобедренного сустава
Врожденная дисплазия тазобедренного сустава - недоразвитие тканей тазобедренного сустава, отсутствие соответствия между суставными поверхностями - состояние, предшествующее вывиху тазобедренного сустава
При дисплазии - применение различных ортезов (приспособлений для отведения бедер) у детей до года. При вывихе - вправление, наложение специальных ортезов в первые месяцы жизни. При безрезультатности такого лечения - хирургическая операция.
Незаращение верхней губы (заячья губа)
Несращение боковых частей верхней губы с ее средней частью. Может быть односторонним и двусторонним. Затрудняет сосание
Хирургическая операция в первые месяцы жизни
Незаращение неба (волчья пасть)
Незаращение верхней челюсти и твердого неба, в результате чего получается расщелина, соединяющая полости рта и носа. Вызывает нарушение питания (попадание пищи в дыхательное горло, в полость носа), дыхания и речи. Часто сочетается с расщелиной в верхней губе
Хирургическая операция и протезирование; диспансерное наблюдение (смена лечебных аппаратов) до 16 лет
Полидактилия - многопалость, наличие лишних пальцев на кисти или стопе. Наиболее частый из врожденных пороков развития; чаще всего встречается в форме шестипалости, обычно на одной конечности.
Врожденный порок сердца
Неправильное внутриутробное формирование перегородки сердца (например, незаращение межпредсердной или межжелудочковой перегородки) либо сохранение после рождения особенностей внутриутробного кровообращения (например, открытый боталлов проток)
При незначительных дефектах межжелудочковой перегородки по мере роста сердца относительный размер отверстия уменьшается - вплоть до полного спонтанного закрытия. В других случаях - хирургическое лечение
Хотелось бы еще раз подчеркнуть, что когда речь идет о врожденных пороках развития, вопрос "Кто виноват?" не только непродуктивен, но и вреден, потому что отвлекает внимание от главного вопроса - "Что делать?". Поговорим на эту тему.
Что делать, если вы планируете беременность
- мужчины и женщины, в чьих семьях уже встречалось то или иное наследственное заболевание, - даже если сами они не больны
- семьи, где уже есть дети, страдающие врожденными пороками развития
- семьи, в которых предыдущие беременности заканчивались выкидышами или мертворождениями
- супруги, состоящие в родстве (например двоюродные и троюродные братья и сестры)
- женщины старше 35 и мужчины старше 50 лет
- мужчины и женщины, в связи со своим родом деятельности, состоянием здоровья или по каким-то иным причинам подвергающиеся воздействию перечисленных выше тератогенных факторов
Во всех этих случаях мы настоятельно рекомендуем партнерам, планирующим беременность, посетить медико-генетическую консультацию. Специалисты-генетики составят родословную, определят риск рождения ребенка с наследственным заболеванием. Нынешний уровень развития медицинских технологий позволяет сегодня в случае неблагоприятного прогноза прибегнуть к искусственному осеменению спермой донора или оплодотворению донорской яйцеклетки. Кроме того, следует по возможности исключить или свести к минимуму воздействие тератогенных факторов.
Что делать, если вы ждете ребенка?
Если вы ждете ребенка и входите в одну из перечисленных "групп риска". Первым шагом и в этом случае должен быть визит в медико-генетическую консультацию. Говорить об этом невесело, но бывают - хотя и очень редко - ситуации, когда на основании одной только родословной генетики приходят к заключению, что плод поражен заболеванием, несовместимым с жизнью. В таком случае, конечно, рекомендуется прерывание беременности. Однако, повторимся, случаи эти очень и очень редки. Как правило, специалисты медико-генетической консультации занимаются не диагностикой, а оценкой риска рождения ребенка с тяжелыми аномалиями и на основании этой оценки рекомендуют тот или иной метод пренатальной диагностики . Далее решение принимается в зависимости от результатов исследования. Насколько на самом деле высок риск родить ребенка с пороками развития, может решить лишь специалист. Не торопитесь делать аборт, если вы прочитали в аннотации, что лекарственный препарат, который вы принимали в самом начале беременности, не рекомендуется использовать в этот период; если принимали алкоголь, наркотики или перенесли острую респираторную вирусную инфекцию, сделали рентгеновский снимок на фоне беременности и т.п. Обязательно обратитесь в медико-генетическую консультацию, где сумеют правильно оценить реальный риск и порекомендуют необходимый комплекс исследований.
Что делать, если у вас родился ребенок с врожденным пороком развития
За окончательной медико-генетической консультацией по поводу прогноза на будущее лучше обратиться через 2-3 месяца, когда спадет психологическая напряженность и супруги смогут более объективно воспринимать такого рода информацию. Для большинства семей последующие беременности бывают успешными. Возможности пренатальной диагностики добавляют уверенности в благополучном исходе и врачам, и пациентам.
Имеются противопоказания. Ознакомьтесь с инструкцией или проконсультируйтесь у специалиста.
Врожденные заболевания являются одной из распространенных причин младенческой и детской смертности, развития хронических болезней у детей и инвалидизации.
Быстрый переход
Врожденные заболевания могут быть обусловлены генетическими мутациями (передающимися по наследству или спонтанными), инфекциями матери во время беременности (цитомегаловирус, ветряная оспа, краснуха), воздействием лекарств и химических веществ, загрязняющих воздух, воду или пищу. Причины множества врожденных дефектов до сих пор неизвестны.
Генетическими или наследственными факторами обусловлены около 20 % врожденных заболеваний. К ним относятся нарушения, при которых мутация затрагивает один ген (серповидно-клеточная анемия); хромосомные нарушения, при которых хромосомы (или их части) отсутствуют (синдром Тернера) или имеют структурные изменения (увеличение количества хромосом или трисомия при синдроме Дауна); многофакторные нарушения, вызванные мутациями двух и более генов. Врожденные дефекты и нарушения развития могут вызывать делеции или дупликации отдельных генов (изменения митохондриальной ДНК). Примером такого заболевания является муковисцидоз, характеризующееся поражением экзокринных желез и жизненно-важных органов (легких и желудочно-кишечного тракта). Врожденные заболевания также ассоциированы со случайными (новыми) повреждениями генов, спонтанными (не наследующимися от родителей) мутациями (большинство случаев ахондроплазии).
По статистике, врожденные заболевания имеют 2–3 % младенцев. К возрасту 1 года их число увеличивается до 5 %, поскольку не все эти патологии диагностируются сразу после рождения.
Врожденные (наследственные) заболевания также классифицируют по типу наследования. При аутосомно-доминантном наследовании заболевание может передаваться от родителя к ребенку в 50 % случаев (мышечная дистрофия Дюшенна, хорея Гентингтона). При аутосомно-рецессивном наследовании генетическая аномалия передается ребенку только в том случае, если оба родителя наделены одним и тем же дефектным геном (муковисцидоз, серповидно-клеточная анемия). Здесь частота наследования составит 25 %, то есть в среднем у 1 ребенка из 4 детей этих родителей будет аутосомно-рецессивное заболевание.
Патологические состояния могут передаваться и при наследовании, сцепленном с полом (наследование гена, находящегося в половых хромосомах). Х-сцепленные рецессивные заболевания почти всегда ограничены мужским полом (гемофилия, дальтонизм, мышечная дистрофия Дюшенна). Х-сцепленные доминантные заболевания встречаются как у мальчиков, так и у девочек, однако у детей мужского пола имеют более тяжелое течение (Х-сцепленный гипофосфатемический рахит). Y-сцепленные заболевания встречаются довольно редко, поскольку Y-хромосома содержит всего несколько генов (ихтиоз).
Некоторые врожденные заболевания формально не относятся к генетическим, но имеют ту или иную выраженность наследования: наследуются факторы риска либо сам ген, но с низкой пенетрантностью (частотой проявления гена в признаках).
Гемофилия
Гемофилия — группа редких наследственных нарушений свертываемости крови, вызванных дефицитом необходимого белка (фактора свертывания крови).
Гемофилия A (классическая) встречается чаще (>80 % случаев) и связана с дефицитом VIII фактора свертывания крови, гемофилия B (болезнь Кристмаса) — реже (>10 % случаев), она обусловлена недостаточностью IX фактора свертывания крови. Гемофилия C встречается очень редко, обусловлена дефицитом XI фактора свертывания крови, чаще всего в классификации группы ее не упоминают.
Заболевание относится к X-сцепленным с полом, наследуется по рецессивному признаку по женской линии. Классическую гемофилию вызывают мутации гена F8, расположенного на X-хромосоме. Примерно в 70 % случаев заболевание наследуется по Х-сцепленному образцу, в остальных случаях оно возникает спонтанно (новая мутация), в дальнейшем спонтанное заболевание становится наследственным. Гемофилией A болеют практически исключительно мужчины, редкие случаи заболевания у женщин, носительниц дефектного гена, почти всегда характеризуются легким течением. Гемофилию B вызывают мутации гена F9, так же расположенного на X-хромосоме, она характерна для мужчин, женщины болеют очень редко. В некоторых случаях заболевание возникает спонтанно (приобретенная гемофилия A или B) и тоже связано с недостаточностью VIII или IX факторов свертывания крови. Приобретенная гемофилия является аутоиммунным заболеванием, при котором организм вырабатывает антитела, атакующие факторы свертывания крови (чаще всего VIII фактор). Примерно в половине случаев приобретенной гемофилии у пациента имеется связанное с ней основное состояние или заболевание (беременность, аутоиммунные заболевания, миелопролиферативные заболевания, воспалительные заболевания кишечника и др.), в остальных эпизодах причина остается невыясненной.
Гемофилия A затрагивает примерно 1 из 5 000 новорожденных мальчиков, гемофилия B — примерно 1 из 25 000. Около 60 % пациентов имеют тяжелую форму гемофилии, обычно им ставят диагноз при рождении или в течение первых 2 лет жизни.
Возраст манифестации гемофилии и тяжесть течения заболевания зависят от уровня активности факторов свертывания крови. Легкая форма характеризуется уровнем фактора свертывания крови (VIII или IX), превышающим 5 % от нормы, средняя — 1–5 %, тяжелая — ниже 1 %. У большинства пациентов, независимо от тяжести течения заболевания, эпизоды кровотечения чаще встречаются в раннем детском, детском и подростковом возрасте, чем в дальнейшей взрослой жизни.
При легкой и умеренной формах гемофилии длительные кровотечения могут возникнуть только в результате травмы, хирургического вмешательства или стоматологической процедуры. Нередко диагноз гемофилии ребенку устанавливают к 5–6 годам, обратив внимание на длительное посттравматическое кровотечение, длительное кровотечение во время стоматологического лечения или операции. К другим заметным симптомам легкого и умеренного течения гемофилии относятся непроходящие гематомы (синяки), частые носовые кровотечения, кровоточивость десен.
Для тяжелой формы гемофилии характерны эпизоды спонтанного кровотечения, которые приводят к кровоизлияниям различной локализации — в мягкие ткани, мышцы, суставы. При гемартрозах (кровоизлияние в полость сустава) возникает ограничение подвижности суставов, сопровождающееся острой болью и воспалением. У пациентов с тяжелой формой гемофилии спонтанные кровотечения чаще всего происходят в мышцы и суставы, однако могут затрагивать любой внутренний орган, включая почки, органы ЖКТ, головной мозг (гематурия, мелена, гематохезия, желудочно-кишечные кровотечения, внутричерепные кровотечения). Тяжелая форма гемофилии A обычно проявляется в раннем детском возрасте, диагноз чаще всего устанавливается к 2 годам ребенка. Обычными симптомами (при отсутствии лечения заболевания) здесь являются кровотечения из-за незначительных травм ротовой полости (прикусывание губ, языка, щек), подкожные гематомы, большие шишки после удара головой, спонтанные кровотечения (2–5 эпизодов в месяц).
Диагноз гемофилии A или B устанавливается на основании симптомов, истории личного и семейного анамнеза пациента, лабораторных исследований (общего анализа крови, коагулограммы, оценивающей состояние системы свертывания крови и активности ее белков, молекулярно-генетического исследования). Парам из группы риска по рождению ребенка с гемофилией необходимо получить генетическую консультацию на этапе планирования беременности. Определение конкретной мутации гена F8 (или F9) помогает не только выявить женщин-носительниц дефектного гена в конкретной семье, но и полезно для пренатальной диагностики гемофилии (амниоцентез, биопсия хориона).
Некоторым пациентам с легкой формой гемофилии назначается синтетический аналог вазопрессина — десмопрессин, повышающий VIII фактор свертывания крови (вводится внутривенно или интраназально), а также антифибринолитические средства, замедляющие распад факторов свертывания крови.
Примерно в 30 % случаев лечения тяжелых случаев гемофилии длительная заместитетельная терапия может приводить к изоиммунизации — образованию антител к вводимым факторам свертывания крови (иммунная система распознает вводимый фактор VIII как чужеродный). Этот процесс может сопровождаться аллергическими реакциями (разной степени тяжести), возрастает риск жизнеугрожающих кровотечений. В этом случае пациенту назначается альтернативное лечение — плазмаферез, иммунодепрессанты.
Гемохроматозы
Гемохроматоз — наследственное заболевание, характеризующееся нарушением обмена железа и его накоплением в тканях и органах.
Заболевание сопровождается повышенным всасыванием железа в желудочно-кишечном тракте и накоплением в печени, сердце, поджелудочной железе, суставах, гипофизе, что приводит к полиорганной недостаточности и развитию таких болезней, как цирроз печени, рак печени, диабет, болезни сердца и суставов.
Наследственный гемохроматоз вызывают мутации генов HFE, HFE2, HAMP, SLC40A1 и TfR2, он классифицируется в зависимости от возраста начала, генетической причины и способа наследования: гемохроматоз 1 типа, гемохроматоз 2 типа, гемохроматоз 3 типа, гемохроматоз 4 типа. Вторичный гемохроматоз ассоциирован с другими заболеваниями (не является наследственным) — анемией, хронической болезнью печени, инфекциями.
Гемохроматоз, редко возникающий в младенческом возрасте и не имеющий явной причины, называют неонатальным гемохроматозом. При этой форме заболевания избыточное накопление железа в тканях и органах начинается еще до рождения ребенка. Неонатальный гемохроматоз быстро прогрессирует и характеризуется поражениями печени, которые выявляют при рождении или в первые дни жизни. Дети с этим заболеванием часто рождаются недоношенными или имеют нарушения внутриутробного развития. Точная причина неонатального гемохроматоза неизвестна, есть версия, что он развивается в том случае, если иммунная система матери распознает клетки печени ребенка как чужеродные. Симптомы обычно включают гипогликемию, нарушения свертываемости крови, пожелтение кожных покровов и склер глаз, отеки. Диагноз устанавливается на основании признаков и симптомов, лабораторных и инструментальных исследований, биопсии печени. Лечение включает переливание крови, внутривенное введение иммуноглобулинов, трансплантацию печени.
Гемохроматоз 1 типа ассоциирован с мутациями в гене HFE, расположенном на коротком плече 6–й хромосомы, наследуется по аутосомно-рецессивному типу, является наиболее распространенным типом наследственного гемохроматоза и поражает в основном мужчин. Вероятность того, что ребенок унаследует мутацию в гене HFE от родителя составляет 25 %, вероятность того, что он станет носителем дефектного гена — 50 %.
Начальные симптомы заболевания обычно отмечаются в возрасте 40–60 лет и включают боль в животе, снижение полового влечения, усталость, слабость, боли в суставах, сухость кожи. В дальнейшем проявляются такие симптомы и осложнения, как изменение пигментации кожи (бронзовая кожа), выпадение волос на голове и туловище, аритмия, кардиомиопатия, хроническая сердечная недостаточность, сахарный диабет, гепатомегалия, цирроз печени, спленомегалия, атрофия яичек, аменорея (у женщин) и другие.
Диагноз устанавливается на основании симптомов заболевания, лабораторных исследований (уровень железа, ферритина, трансферрина, железосвязывающей способности сыворотки крови, маркеры функции печени, уровень глюкозы крови и др.), инструментальных исследований (рентгенография суставов, электрокардиография и эхокардиография сердца, УЗИ органов брюшной полости, МРТ печени и др.), биопсии печени, молекулярно-генетического исследования (в том числе для выявления родственников — имеющих гемохроматоз или носителей).
Лечение гемохроматоза направлено на удаление избытка железа из организма (флеботомия, хелатирование) с помощью забора крови (как при взятии анализов или донорстве крови, только в большем объеме) и специальных препаратов, образующих комплексное соединение с железом и способствующих его удалению из железосодержащих белков (дефероксамин).
Терапевтическая флеботомия сначала проводится 1–2 раза в неделю, поддерживающая — каждые 2–4 месяца. Вместе с этим проводится симптоматическое лечение сахарного диабета, патологий сердца, цирроза печени и других заболеваний, вызванных гемохроматозом. Пациентам запрещен прием препаратов железа и любых медикаментозных средств или комплексов, которые могут содержать железо и витамин C, запрещен алкоголь во избежание дальнейшего повреждения печени (если оно имеется), рекомендуется придерживаться диеты, исключающей продукты с высоким содержанием железа (красное мясо, яблоки, печень, шпинат).
Гемохроматоз 2 типа вызывают мутации в генах HFE2 или HAMP, он наследуется по аутосомно-рецессивному типу и проявляется чаще всего в детском возрасте. Для этого типа заболевания характерны следующие симптомы: высокие уровни ферритина и трансферрина, врожденный фиброз печени, артропатия, кардиомиопатия, генерализованная гиперпигментация кожи, мышечная слабость, гипогонадизм, задержка полового созревания, сахарный диабет, цирроз печени, остеопороз, спленомегалия, гепатомегалия и другие. Диагностика и лечение гемохроматоза 2 типа проводятся аналогично диагностике и лечению гемохроматоза 1 типа.
Гемохроматоз 3 типа вызывают мутации в гене TFR2, он наследуется по аутосомно-рецессивному типу. Симптомы обычно проявляются до 30 лет и, помимо вышеперечисленных, могут включать снижение уровня лимфоцитов и нейтрофилов в крови, красные или пурпурные пятна на коже. Диагностика и лечение гемохроматоза 3 типа проводятся аналогично диагностике и лечению гемохроматоза 1 и 2 типов.
Симптомы гемохроматоза 4 типа могут проявиться как в детском, так и во взрослом возрасте. Этот тип заболевания вызывают мутации гена SLC40A1, наследуется оно по аутосомно-доминатному типу. Вероятность того, что ребенок унаследует мутацию в гене SLC40A1 от родителя составляет 50 %, соответственно, вероятность того, что он станет носителем дефектного гена — тоже 50 %. Симптомы гемохроматоза 4 типа могут проявляться в виде повышенной утомляемости, слабости, болей в суставах, изменения пигментации кожи, затруднения дыхания, сердечной недостаточности, сахарного диабета, анемии, врожденного фиброза печени, цирроза печени, остеоартроза, катаракты. Диагностика и лечение гемохроматоза 4 типа проводятся аналогично диагностике и лечению гемохроматоза 1, 2 и 3 типов.
Наследственный гемохроматоз — заболевание с пониженной пенетрантностью (вероятностью того, что ген будет иметь любые фенотипические проявления) дефектного гена, т. е. у некоторых людей с мутациями гена HFE никогда не проявятся симптомы, при этом у их детей или других членов семьи с мутацией гена произойдет манифестация заболевания.
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна — редкое наследственное заболевание, характеризующееся нарастающей мышечной слабостью с последующей атрофией мышц. Ассоциировано с мутацией гена DMD, расположенного на половой X-хромосоме (X-сцепленный рецессивный тип наследования).
Наследственные заболевания сетчатки — клинически и генетически гетерогенная группа состояний: многие из них манифестируют в детском возрасте как изолированная аномалия у практически здорового ребенка. Некоторые развиваются на фоне системных аномалий. Идентифицировано большинство генов, вызывающих основные дистрофии сетчатки у детей, выявлены сложные связи генотипа с фенотипом. Отмечается выраженная генетическая гетерогенность отдельных случаев генетических заболеваний, мутация одного гена может вызывать развитие нескольких разных фенотипов. Несмотря на это, данные заболевания можно классифицировать по следующим признакам:
1. Стационарные или прогрессирующие
2. Проявляются поражением преимущественно палочек или колбочек.
Стационарные заболевания манифестируют при рождении или в первые месяцы жизни и наиболее точно описываются как синдромы дисфункции. Прогрессирующие состояния, как правило, проявляющиеся позже, называются дистрофиями.
Стационарные синдромы дисфункции сетчатки группа заболеваний включает в себя различные формы стационарной ночной слепоты (синдромы дисфункции палочек) и синдромы дисфункции колбочек (стационарные болезни колбочек).
Выделяют три основных формы стационарной ночной слепоты; при врожденной стационарной ночной слепоте (congenital stationary night-blindness — CSNB) глазное дно интактно или развиваются изменения, характерные для миопии. При белоточечном глазном дне
а) Клинические проявления. Врожденная стационарная ночная слепота характеризуется ночной слепотой, ухудшением зрения различной степени и отсутствием изменений на глазном дне. Она может наследоваться по аутосомно-доминантному (autosomal dominant — AD), аутосомно-рецессивному (autosomal recessive— AR) или Х-сцепленному механизму (X-linked — XL).
Острота зрения при аутосомно-доминантной форме обычно в пределах нормы или немного снижена, тогда как при аутосомно-рецессивной и Х-сцепленной формах отмечается небольшое или умеренное снижение остроты центрального зрения. Другие проявления Х-сцепленной и аутосомно-рецессивной формы включают в себя близорукость средней или высокой степени, нистагм, косоглазие и парадоксальные реакции зрачков. При осмотре глазного дна патологических изменений обычно не выявляется, у некоторых пациентов могут отмечаться изменения, характерные для близорукости, бледность или наклон диска зрительного нерва.
У пациентов с аутосомно-доминантной врожденной стационарной ночной слепотой заболевание обычно проявляется симптоматической никталопией, но при Х-сцепленной и аутосомно-рецессивной форме болезнь обычно проявляется в младенческом возрасте нистагмом, косоглазием и ухудшением зрения. Нистагм присутствует не у всех пациентов, что приводит к поздней диагностике заболевания в детском или даже взрослом возрасте. Без выполнения электроретинографии (ЭРГ) заболевание может остаться недиагностированным. Х-сцепленный и аутосомно-рецессивный варианты далее можно подразделять на полные и неполные формы. Сначала такая дифференцировка на основании электрофизиологических и психофизиологических критериев была предложена для Х-сцепленной формы, в последствии было показано, что эта классификация разделяет два генетически различных заболевания.
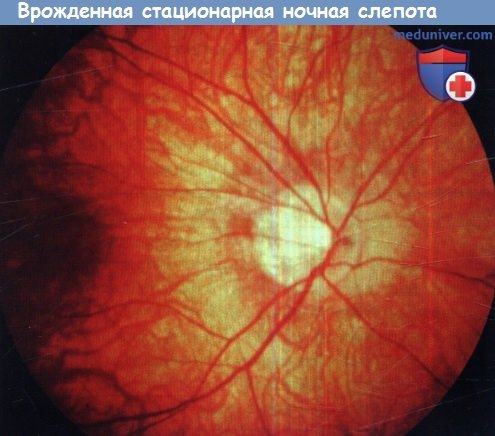
Х-сцепленная врожденная стационарная ночная слепота.
Наклонный диск зрительного нерва и миопические изменения глазного дна.
б) Электрофизиология врожденной постоянной ночной слепоты. Электроретинографию следует выполнять по стандартам Международного общества клинической электрофизиологии зрения (International Society for Clinical Electrophysiology of Vision — ISCEV). При обследовании младенцев исследование может быть невыполнимым, в таких случаях используется модифицированный протокол. Определяется четыре основных ответа: палочковая ЭРГ и ответ на яркую вспышку в скотопических условиях, и два показателя функции колбочек, ритмическая ЭРГ при стимуляции мерцанием 30 Гц и ЭРГ при стимуляции одной вспышкой в фотопических условиях.
На колбочковой ЭРГ фиксируются незначительные аномалии, отражающие дисфункцию биполярных ON-клеток. При неполной врожденной стационарной ночной слепоте генерируется палочковая ЭРГ и глубоко негативный ответ на яркую вспышку. Колбочковая ЭРГ изменена гораздо больше, чем при полной форме врожденной стационарной ночной слепоты, что свидетельствует о поражении как ON-, так и OFF-биполярных клеток. При стимуляции мерцанием регистрируется характерный трехфазный ответ.
При аутосомно-доминантной форме врожденной стационарной ночной слепоты могут возникать изменения, указывающие на дисфункцию системы внутренних элементов системы палочек, но в сочетании с нормальной колбочковой ЭРГ по протоколам ISCEV. В других случаях аутосомно-доминантной формы заболевания ответы на палочковой ЭРГ ослаблены, колбочковые ответы в пределах нормы, но стимуляция стандартной яркой вспышкой не вызывает появления негативной волны.
в) Молекулярная генетика и патогенез врожденной стационарной ночной слепоты:
1. Аутосомно-доминантная врожденная стационарная ночная слепота. При аутосомно-доминантной врожденной стационарной ночной слепоте описаны мутации генов, кодирующих три специфических компонента каскада фототрансдукции палочек: родопсин, а-субъединицу трансдуцина палочек и р-субъединицу фосфодиэстеразы (phosphodiesterase (3-subunit—PDE Р) циклического гуанозин монофосфата (cyclic guanosine monophosphate — cGMP) палочек.
2. Х-сцепленная врожденная стационарная ночная слепота. Идентифицировано два ответственных гена (CACNA1F и NYX), их дефекты вызывают заболевание в большинстве семей. Неполная врожденная стационарная ночная слепота развивается на фоне мутации СACNА1F, кодирующего специфическую a1F-субъединицу вольтаж-зависимого кальциевого канала L-типа сетчатки. Экспрессия CACNA1F, как оказалось, ограничена фоторецепторами, и выражена в синаптических окончаниях. Большинство мутаций представляют собою варианты последовательностей, вызывающих преждевременную терминацию синтеза белка. Недостаточность функционирующих каналов палочек и колбочек нарушает поступление в фоторецепторы кальция, необходимого для тонического выброса нейромедиатора из пресинаптических окончаний.
Это делает невозможным поддержание нормального трансмембранного потенциала биполярных клеток, таким образом, при низкой освещенности сетчатка не способна реагировать на изменения освещенности.
Полная форма врожденной стационарной ночной слепоты вызывается мутацией NYX, гена, кодирующего богатый лейцином протеогликан никталопии. Богатые лейцином последовательности, как считается, играют важную роль во взаимодействии белков, в пределах этих последовательностей идентифицировано множество мутаций. Никталопии экспрессируется во внутреннем сегменте фоторецепторов, наружном и внутреннем ядерном слое и ганглиозными клетками. Возможно, никталопии регулирует и стимулирует формирование и работу ON-путей сетчатки.
На пациентах с мутациями CACNA1F и NYX проведено несколько исследований отношений генотип-фенотип. При мутациях CACNA1F отмечена выраженная межсемейная и внутрисемейная фенотипическая вариабельность, даже при идентичных вариантах последовательностей, что свидетельствует о влиянии на фенотип других генетических факторов и факторов среды. Хотя у большинства пациентов с Х-сцепленной врожденной стационарной ночной слепотой заболевание не прогрессировало, Nakamura et al. описали двух братьев с мутацией CACNA1F, прогрессирующим ухудшением зрения и, в терминальной стадии заболевания, нерегистрируемыми палочковой и колбочковой ЭРГ. Изредка мы также наблюдали медленное прогрессирование заболевания у пациентов с Х-сцепленной врожденной стационарной ночной слепотой.
Пациенты с полной врожденной стационарной ночной слепотой (мутации NYX) всегда страдают миопией и гораздо более выраженной никталопией.
3. Аутосомно-рецессивная врожденная стационарная ночная слепота. Мутации GRM6 и TRPM1 вызывают полную форму врожденной стационарной ночной слепоты. GRM6 кодирует метаботропный глутаматный рецептор (mGluR6) дендритов палочек, колбочек и ON-биполярных клеток, участвующий в перемене знака (потенциала) в первом синапсе, таким образом, выброс фоторецепторами в темноте глутамата вызывает гиперполяризацию мембраны ON-биполярной клетки. TRPM1, транзиторный рецепторный потенциал-зависимый катионный канал, подсемейство М, представитель 1, вероятно, влияет на изменения потенциала мембраны ON-биполярных клеток в ответ на воздействие глутамата.
Мутации САВР4 вызывают неполную форму врожденной стационарной ночной слепоты. САВР4, относящийся к семейству кальций-связывающих белков (calcium-binding protein — САВР), локализуется исключительно на синаптических окончаниях фоторецепторов, где он напрямую связан с С-концевым доменом CACNA1F.
У пациентов с аутосомно-рецессивной врожденной стационарной ночной слепотой при отсутствии негативной ЭРГ были идентифицированы варианты последовательности SLC24A1; при стандартной стимуляции яркой вспышкой в скотопических условиях регистрировалось одинаковое уменьшение амплитуды а- и b-волн. SLC24A1 относится к надсемейству белков — переносчиков растворенных веществ и локализуется во внутренних сегментах (фоторецепторов), наружном и внутреннем ядерных слоях и ганглиозных клетках.
4. Офтальмопатия Аландских островов. Офтальмопатия Аландских островов (Aland Island eye disease — AIED) — Х-сцепленное рецессивное заболевание, сходное с неполной врожденной стационарной ночной слепотой, характеризующееся снижением остроты зрения, нистагмом, никталопией, легкой красно-зеленой дисхроматопсией и близорукостью. У больных мужчин может наблюдаться просвечивающая радужка, гипоплазия центральной ямки и гипопигментация глазного дна. Клиническая картина может напоминать картину Х-сцепленного глазного альбинизма (X-linked ocular albinism — XLOA), но при Х-сцепленном глазном альбинизме цветовосприятие обычно в норме, а у пациентов с офтальмопатией Аландских островов не выявляется характерная для альбинизма аномалия нервных волокон хиазмы.
Ночная слепота, психофизические изменения и изменения на ЭРГ при офтальмопатии Аландских островов аналогичны наблюдаемым при неполной форме Х-сцепленной врожденной стационарной ночной слепоты. Оба заболевания картированы в одной зоне Хр: вероятно, они аллельны друг другу, но мутации CACNA1F при офтальмопатии Аландских островов выявлены не были.
5. Другие близкие фенотипы. Пациенты с синдромами, вызываемыми аномалиями соседних генов (включающих в себя недостаточность глицеролкиназы, врожденную гипоплазию надпочечников, мышечную дистрофию Дюшенна (Duchenne’s muscular dystrophy — DMD) и аномалии глаз, известные как орегонская офтальмопатия) и делецией Хр21, имеют те же дефекты глаз, что и мужчины, больные офтальмопатией Аландских островов и те же изменения на ЭРГ, указывающие на поражение преимущественно внутренних слоев сетчатки. Более того, у некоторых мужчин с изолированной мышечной дистрофией Дюшенна (мутацией гена дистрофина на Хр21) регистрируются те же изменения ЭРГ, что и при врожденной стационарной ночной слепоте. Все эти мультисистемные расстройства сопровождаются непрогрессирующей дисфункцией сетчатки, преимущественно палочек.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Дальтонизм – наверное, одно из самых известных офтальмологических заболеваний: ему посвящены и анекдоты, и афоризмы. При этом чаще всего они не отражают сути заболевания: поэтому в сознании людей оно часто ассоциируется с несвойственными для него симптомами и особенностями.
О том, что же на самом деле представляет собой дальтонизм, каковы его природа и механизмы, какие бывают виды дальтонизма и есть ли способы его вылечить – в нашей статье.
Дальтонизм – что это такое
Дальтонизм – или цветовая слепота – это состояние, при котором частично или полностью теряется возможность различать цвета. Степень аномалии варьируется – для некоторых ограничения касаются всего одного цвета, а кто-тоже видит мир черно-белым.
Вопреки распространенному мнению, восприятие дальтоников не подменяет один цвет другим: они не видят желтые цветы синими, а красный зонтик – голубым. В их картине мира некоторые цвета как бы сливаются, приближаются друг к другу, другие же остаются сохранными. Конечно, цветовосприятиебудет зависеть от типов дефекта, о которых мы поговорим ниже.
Феномен дальтонизма назван в честь английского метеоролога, химика, физика Джона Дальтона, жившего в 18 веке. Только в 26 лет ученый вдруг понял, что воспринимает краски иначе, чем остальные: причем это открытие подарил ему собственный пиджак. Дальтон был твердо уверен, что используемая им деталь гардероба – серого цвета, а на самом деле одежда была темно-красной. Ученый развил тему и на основании своих ощущений описал, как страдающие от той же особенности воспринимают мир.
Правильнее называть дальтонизм не заболеванием, а особенностью организма или дефектом зрения: он не представляет угрозы здоровью пациента. Их отличие от обычных людей заключается лишь в том, что некоторые цвета – и, соответственно, общую картину окружающего мира – они видят иначе.

Механизм аномалии
Формы и виды дальтонизма
В некоторых случаях в колбочках могут быть представлены все три пигмента, однако активность одного из них понижена. В этом случае речь идет об аномальной трихромазии.
Аномальная трихромазия бывает трёх видов:
Еще один вариант – отсутствие одного из трех пигментов (чаще – красного, гораздо реже – синего). Такое состояние называется дихромазией.
В зависимости от того, какой из пигментов отсутствует, выделяют:
- протанопию (когда нет красного пигмента);
- дейтеранопию (отсутствует зеленый пигмент);
- тританопию (не хватает синего пигмента).

Самым редким проявлением заболевания считается полное отсутствие в колбочках всех трех пигментов – монохромазия или ахромазия.
Причины дальтонизма
Дальтонизм – достаточно распространенное состояние: по статистике каждый восьмой мужчина сталкивается с нарушением цветовосприятия. Вероятность дальтонизма у женщин гораздо меньше: это случается всего в 0,4 случаях из 100.
Причины дальтонизма могут быть генетическими или приобретенными.
Ген дальтонизма сцеплен с X-хромосомой: аномалия передается по женской линии. Если мама ребенка – дальтоник, ее сын унаследует ту же особенность, а дочь станет носительницей и передаст своим детям. В тех редких случаях, когда оба родителя – дальтоники, ребенок будет страдать от дефектов цветовосприятия. Феномен, объясняющий, как наследуется признак дальтонизма, называется сцепленным наследованием.
Полная цветовая слепота может быть только наследственной. Частота ее проявления – 1 случай на миллион.
Кроме того, генетический дальтонизм может быть связан с такими патологиями как дистрофия колбочек и амавроз Лебера.
Приобретенная форма дальтонизма связана с травмами, старением, заболеваниями зрительного нерва, диабетом. В этом случае аномалия затронет только пораженный глаз и будет иметь тенденцию к ухудшению.
Диагностика дальтонизма
При подозрении на нарушения цветовосприятия офтальмологи пользуются специальными таблицами, аномалоскопами Нагеля и Рабкина, мерцательными фонарями и другими методами.
Полихроматические таблицы (Ишихара, Юстовой, Рабкина) выглядят как организованное скопление разноразмерных кружков основного и дополнительного цветов одинаковой яркости. Часть кружков образовывают на фоне остальных извивающуюся линию, фигуру или цифру. Некоторые таблицы позволяют не только выявить дефект, но и определить тип дальтонизма и его степень.

Метод Хольмгрена. Пациенту предлагают выбрать из 133 мотков разноцветной шерсти такие, которые относятся к одному цвету, но разным оттенкам.
Коррекция дальтонизма

Если же патология стала следствием повреждения головного мозга, для лечения может быть назначено хирургическое вмешательство.
Для коррекции можно использовать специальную оптику – очки или линзы, которая, усиливает восприятие основных цветов, приближая зрение дальтоника к нормальному. Существуют также специальные высокотехнологичные кибернетические приспособления и программное обеспечение, помогающее дальтоникам при работе. Однако полностью превратить дальтонизм в нормальное зрение не получится.
Читайте также: