
К какому виду наследования относятся все заболевания глаз
Обновлено: 02.07.2024
Врожденные заболевания являются одной из распространенных причин младенческой и детской смертности, развития хронических болезней у детей и инвалидизации.
Быстрый переход
Врожденные заболевания могут быть обусловлены генетическими мутациями (передающимися по наследству или спонтанными), инфекциями матери во время беременности (цитомегаловирус, ветряная оспа, краснуха), воздействием лекарств и химических веществ, загрязняющих воздух, воду или пищу. Причины множества врожденных дефектов до сих пор неизвестны.
Генетическими или наследственными факторами обусловлены около 20 % врожденных заболеваний. К ним относятся нарушения, при которых мутация затрагивает один ген (серповидно-клеточная анемия); хромосомные нарушения, при которых хромосомы (или их части) отсутствуют (синдром Тернера) или имеют структурные изменения (увеличение количества хромосом или трисомия при синдроме Дауна); многофакторные нарушения, вызванные мутациями двух и более генов. Врожденные дефекты и нарушения развития могут вызывать делеции или дупликации отдельных генов (изменения митохондриальной ДНК). Примером такого заболевания является муковисцидоз, характеризующееся поражением экзокринных желез и жизненно-важных органов (легких и желудочно-кишечного тракта). Врожденные заболевания также ассоциированы со случайными (новыми) повреждениями генов, спонтанными (не наследующимися от родителей) мутациями (большинство случаев ахондроплазии).
По статистике, врожденные заболевания имеют 2–3 % младенцев. К возрасту 1 года их число увеличивается до 5 %, поскольку не все эти патологии диагностируются сразу после рождения.
Врожденные (наследственные) заболевания также классифицируют по типу наследования. При аутосомно-доминантном наследовании заболевание может передаваться от родителя к ребенку в 50 % случаев (мышечная дистрофия Дюшенна, хорея Гентингтона). При аутосомно-рецессивном наследовании генетическая аномалия передается ребенку только в том случае, если оба родителя наделены одним и тем же дефектным геном (муковисцидоз, серповидно-клеточная анемия). Здесь частота наследования составит 25 %, то есть в среднем у 1 ребенка из 4 детей этих родителей будет аутосомно-рецессивное заболевание.
Патологические состояния могут передаваться и при наследовании, сцепленном с полом (наследование гена, находящегося в половых хромосомах). Х-сцепленные рецессивные заболевания почти всегда ограничены мужским полом (гемофилия, дальтонизм, мышечная дистрофия Дюшенна). Х-сцепленные доминантные заболевания встречаются как у мальчиков, так и у девочек, однако у детей мужского пола имеют более тяжелое течение (Х-сцепленный гипофосфатемический рахит). Y-сцепленные заболевания встречаются довольно редко, поскольку Y-хромосома содержит всего несколько генов (ихтиоз).
Некоторые врожденные заболевания формально не относятся к генетическим, но имеют ту или иную выраженность наследования: наследуются факторы риска либо сам ген, но с низкой пенетрантностью (частотой проявления гена в признаках).
Гемофилия
Гемофилия — группа редких наследственных нарушений свертываемости крови, вызванных дефицитом необходимого белка (фактора свертывания крови).
Гемофилия A (классическая) встречается чаще (>80 % случаев) и связана с дефицитом VIII фактора свертывания крови, гемофилия B (болезнь Кристмаса) — реже (>10 % случаев), она обусловлена недостаточностью IX фактора свертывания крови. Гемофилия C встречается очень редко, обусловлена дефицитом XI фактора свертывания крови, чаще всего в классификации группы ее не упоминают.
Заболевание относится к X-сцепленным с полом, наследуется по рецессивному признаку по женской линии. Классическую гемофилию вызывают мутации гена F8, расположенного на X-хромосоме. Примерно в 70 % случаев заболевание наследуется по Х-сцепленному образцу, в остальных случаях оно возникает спонтанно (новая мутация), в дальнейшем спонтанное заболевание становится наследственным. Гемофилией A болеют практически исключительно мужчины, редкие случаи заболевания у женщин, носительниц дефектного гена, почти всегда характеризуются легким течением. Гемофилию B вызывают мутации гена F9, так же расположенного на X-хромосоме, она характерна для мужчин, женщины болеют очень редко. В некоторых случаях заболевание возникает спонтанно (приобретенная гемофилия A или B) и тоже связано с недостаточностью VIII или IX факторов свертывания крови. Приобретенная гемофилия является аутоиммунным заболеванием, при котором организм вырабатывает антитела, атакующие факторы свертывания крови (чаще всего VIII фактор). Примерно в половине случаев приобретенной гемофилии у пациента имеется связанное с ней основное состояние или заболевание (беременность, аутоиммунные заболевания, миелопролиферативные заболевания, воспалительные заболевания кишечника и др.), в остальных эпизодах причина остается невыясненной.
Гемофилия A затрагивает примерно 1 из 5 000 новорожденных мальчиков, гемофилия B — примерно 1 из 25 000. Около 60 % пациентов имеют тяжелую форму гемофилии, обычно им ставят диагноз при рождении или в течение первых 2 лет жизни.
Возраст манифестации гемофилии и тяжесть течения заболевания зависят от уровня активности факторов свертывания крови. Легкая форма характеризуется уровнем фактора свертывания крови (VIII или IX), превышающим 5 % от нормы, средняя — 1–5 %, тяжелая — ниже 1 %. У большинства пациентов, независимо от тяжести течения заболевания, эпизоды кровотечения чаще встречаются в раннем детском, детском и подростковом возрасте, чем в дальнейшей взрослой жизни.
При легкой и умеренной формах гемофилии длительные кровотечения могут возникнуть только в результате травмы, хирургического вмешательства или стоматологической процедуры. Нередко диагноз гемофилии ребенку устанавливают к 5–6 годам, обратив внимание на длительное посттравматическое кровотечение, длительное кровотечение во время стоматологического лечения или операции. К другим заметным симптомам легкого и умеренного течения гемофилии относятся непроходящие гематомы (синяки), частые носовые кровотечения, кровоточивость десен.
Для тяжелой формы гемофилии характерны эпизоды спонтанного кровотечения, которые приводят к кровоизлияниям различной локализации — в мягкие ткани, мышцы, суставы. При гемартрозах (кровоизлияние в полость сустава) возникает ограничение подвижности суставов, сопровождающееся острой болью и воспалением. У пациентов с тяжелой формой гемофилии спонтанные кровотечения чаще всего происходят в мышцы и суставы, однако могут затрагивать любой внутренний орган, включая почки, органы ЖКТ, головной мозг (гематурия, мелена, гематохезия, желудочно-кишечные кровотечения, внутричерепные кровотечения). Тяжелая форма гемофилии A обычно проявляется в раннем детском возрасте, диагноз чаще всего устанавливается к 2 годам ребенка. Обычными симптомами (при отсутствии лечения заболевания) здесь являются кровотечения из-за незначительных травм ротовой полости (прикусывание губ, языка, щек), подкожные гематомы, большие шишки после удара головой, спонтанные кровотечения (2–5 эпизодов в месяц).
Диагноз гемофилии A или B устанавливается на основании симптомов, истории личного и семейного анамнеза пациента, лабораторных исследований (общего анализа крови, коагулограммы, оценивающей состояние системы свертывания крови и активности ее белков, молекулярно-генетического исследования). Парам из группы риска по рождению ребенка с гемофилией необходимо получить генетическую консультацию на этапе планирования беременности. Определение конкретной мутации гена F8 (или F9) помогает не только выявить женщин-носительниц дефектного гена в конкретной семье, но и полезно для пренатальной диагностики гемофилии (амниоцентез, биопсия хориона).
Некоторым пациентам с легкой формой гемофилии назначается синтетический аналог вазопрессина — десмопрессин, повышающий VIII фактор свертывания крови (вводится внутривенно или интраназально), а также антифибринолитические средства, замедляющие распад факторов свертывания крови.
Примерно в 30 % случаев лечения тяжелых случаев гемофилии длительная заместитетельная терапия может приводить к изоиммунизации — образованию антител к вводимым факторам свертывания крови (иммунная система распознает вводимый фактор VIII как чужеродный). Этот процесс может сопровождаться аллергическими реакциями (разной степени тяжести), возрастает риск жизнеугрожающих кровотечений. В этом случае пациенту назначается альтернативное лечение — плазмаферез, иммунодепрессанты.
Гемохроматозы
Гемохроматоз — наследственное заболевание, характеризующееся нарушением обмена железа и его накоплением в тканях и органах.
Заболевание сопровождается повышенным всасыванием железа в желудочно-кишечном тракте и накоплением в печени, сердце, поджелудочной железе, суставах, гипофизе, что приводит к полиорганной недостаточности и развитию таких болезней, как цирроз печени, рак печени, диабет, болезни сердца и суставов.
Наследственный гемохроматоз вызывают мутации генов HFE, HFE2, HAMP, SLC40A1 и TfR2, он классифицируется в зависимости от возраста начала, генетической причины и способа наследования: гемохроматоз 1 типа, гемохроматоз 2 типа, гемохроматоз 3 типа, гемохроматоз 4 типа. Вторичный гемохроматоз ассоциирован с другими заболеваниями (не является наследственным) — анемией, хронической болезнью печени, инфекциями.
Гемохроматоз, редко возникающий в младенческом возрасте и не имеющий явной причины, называют неонатальным гемохроматозом. При этой форме заболевания избыточное накопление железа в тканях и органах начинается еще до рождения ребенка. Неонатальный гемохроматоз быстро прогрессирует и характеризуется поражениями печени, которые выявляют при рождении или в первые дни жизни. Дети с этим заболеванием часто рождаются недоношенными или имеют нарушения внутриутробного развития. Точная причина неонатального гемохроматоза неизвестна, есть версия, что он развивается в том случае, если иммунная система матери распознает клетки печени ребенка как чужеродные. Симптомы обычно включают гипогликемию, нарушения свертываемости крови, пожелтение кожных покровов и склер глаз, отеки. Диагноз устанавливается на основании признаков и симптомов, лабораторных и инструментальных исследований, биопсии печени. Лечение включает переливание крови, внутривенное введение иммуноглобулинов, трансплантацию печени.
Гемохроматоз 1 типа ассоциирован с мутациями в гене HFE, расположенном на коротком плече 6–й хромосомы, наследуется по аутосомно-рецессивному типу, является наиболее распространенным типом наследственного гемохроматоза и поражает в основном мужчин. Вероятность того, что ребенок унаследует мутацию в гене HFE от родителя составляет 25 %, вероятность того, что он станет носителем дефектного гена — 50 %.
Начальные симптомы заболевания обычно отмечаются в возрасте 40–60 лет и включают боль в животе, снижение полового влечения, усталость, слабость, боли в суставах, сухость кожи. В дальнейшем проявляются такие симптомы и осложнения, как изменение пигментации кожи (бронзовая кожа), выпадение волос на голове и туловище, аритмия, кардиомиопатия, хроническая сердечная недостаточность, сахарный диабет, гепатомегалия, цирроз печени, спленомегалия, атрофия яичек, аменорея (у женщин) и другие.
Диагноз устанавливается на основании симптомов заболевания, лабораторных исследований (уровень железа, ферритина, трансферрина, железосвязывающей способности сыворотки крови, маркеры функции печени, уровень глюкозы крови и др.), инструментальных исследований (рентгенография суставов, электрокардиография и эхокардиография сердца, УЗИ органов брюшной полости, МРТ печени и др.), биопсии печени, молекулярно-генетического исследования (в том числе для выявления родственников — имеющих гемохроматоз или носителей).
Лечение гемохроматоза направлено на удаление избытка железа из организма (флеботомия, хелатирование) с помощью забора крови (как при взятии анализов или донорстве крови, только в большем объеме) и специальных препаратов, образующих комплексное соединение с железом и способствующих его удалению из железосодержащих белков (дефероксамин).
Терапевтическая флеботомия сначала проводится 1–2 раза в неделю, поддерживающая — каждые 2–4 месяца. Вместе с этим проводится симптоматическое лечение сахарного диабета, патологий сердца, цирроза печени и других заболеваний, вызванных гемохроматозом. Пациентам запрещен прием препаратов железа и любых медикаментозных средств или комплексов, которые могут содержать железо и витамин C, запрещен алкоголь во избежание дальнейшего повреждения печени (если оно имеется), рекомендуется придерживаться диеты, исключающей продукты с высоким содержанием железа (красное мясо, яблоки, печень, шпинат).
Гемохроматоз 2 типа вызывают мутации в генах HFE2 или HAMP, он наследуется по аутосомно-рецессивному типу и проявляется чаще всего в детском возрасте. Для этого типа заболевания характерны следующие симптомы: высокие уровни ферритина и трансферрина, врожденный фиброз печени, артропатия, кардиомиопатия, генерализованная гиперпигментация кожи, мышечная слабость, гипогонадизм, задержка полового созревания, сахарный диабет, цирроз печени, остеопороз, спленомегалия, гепатомегалия и другие. Диагностика и лечение гемохроматоза 2 типа проводятся аналогично диагностике и лечению гемохроматоза 1 типа.
Гемохроматоз 3 типа вызывают мутации в гене TFR2, он наследуется по аутосомно-рецессивному типу. Симптомы обычно проявляются до 30 лет и, помимо вышеперечисленных, могут включать снижение уровня лимфоцитов и нейтрофилов в крови, красные или пурпурные пятна на коже. Диагностика и лечение гемохроматоза 3 типа проводятся аналогично диагностике и лечению гемохроматоза 1 и 2 типов.
Симптомы гемохроматоза 4 типа могут проявиться как в детском, так и во взрослом возрасте. Этот тип заболевания вызывают мутации гена SLC40A1, наследуется оно по аутосомно-доминатному типу. Вероятность того, что ребенок унаследует мутацию в гене SLC40A1 от родителя составляет 50 %, соответственно, вероятность того, что он станет носителем дефектного гена — тоже 50 %. Симптомы гемохроматоза 4 типа могут проявляться в виде повышенной утомляемости, слабости, болей в суставах, изменения пигментации кожи, затруднения дыхания, сердечной недостаточности, сахарного диабета, анемии, врожденного фиброза печени, цирроза печени, остеоартроза, катаракты. Диагностика и лечение гемохроматоза 4 типа проводятся аналогично диагностике и лечению гемохроматоза 1, 2 и 3 типов.
Наследственный гемохроматоз — заболевание с пониженной пенетрантностью (вероятностью того, что ген будет иметь любые фенотипические проявления) дефектного гена, т. е. у некоторых людей с мутациями гена HFE никогда не проявятся симптомы, при этом у их детей или других членов семьи с мутацией гена произойдет манифестация заболевания.
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна — редкое наследственное заболевание, характеризующееся нарастающей мышечной слабостью с последующей атрофией мышц. Ассоциировано с мутацией гена DMD, расположенного на половой X-хромосоме (X-сцепленный рецессивный тип наследования).
Наследственные заболевания в структуре офтальмопатологии составляют не менее 30%. Одной из наиболее распространенных нозологических форм является наследственная катаракта. При диагностике врачом-офтальмологом учитываются результаты комплексного клинико-лабораторного и параклинического узкоспециализированного обследования, однако при выставлении диагноза редко учитывается этиологический фактор болезни. Молекулярно-генетические исследования позволяют установить истинную причину и патогенез патологического процесса наследственного заболевания. Изолированные наследственные катаракты – это группа заболеваний, обусловленных выраженным клиническим полиморфизмом и генетической гетерогенностью. Эти особенности являются основными причинами затруднения диагностики клинико-генетической формы болезни. Согласно литературным данным, выделяют более 50 генетических форм изолированных наследственных катаракт. Клинико-диагностический алгоритм у пациентов с данной патологией является полноценным только в случае наличия медико-генетического консультирования семей врачами-генетиками с учетом данных офтальмологического обследования и молекулярно-генетического исследования. С учетом верификации этиологической компоненты патологического процесса появляется возможность корректно планировать профилактические и лечебные мероприятия. В настоящее время основным методом лечения катаракты является хирургическое лечение. Функциональное состояние органа зрения, стадия патологического процесса и соматическое состояние пациента, прогноз лечения диктуют корректировку лечебных мероприятий с отменой в некоторых случаях хирургического вмешательства при катаракте и с проведением консервативной терапии. В настоящее время терапия помутнения хрусталика проводится путем применения препаратов метаболического воздействия в виде глазных капель. Пиреноксин 75 мг в виде глазных капель (Каталин) – один из известных препаратов данной группы, он используется в практической офтальмологии 12 стран мира. Каталин показан при возрастной катаракте со слабой или средней степенью интенсивности помутнения хрусталиковых масс.
Ключевые слова: катаракта, наследственные болезни, медицинская генетика, клинический полиморфизм, гетерогенность, профилактика, лечение, пиреноксин.
Clinical and genetic aspects of isolated hereditary cataracts
Kadyshev V.V. 1, Zinchenko R.A. 1,2, Kutsev S.I. 1,2
1 Research Centre for Medical Genetics, Moscow
2 Russian National Research Medical University named after N.I. Pirogov, Moscow
Hereditary diseases in ophthalmology account for at least 30% of cases among the whole structure of ophthalmopathology. One of the most common nosological units is hereditary cataract. Using the diagnostic algorithm the ophthalmologist takes into account the results of a complex clinico-laboratory and highly specialized paraclinical examination, however, when the diagnosis is made, the etiological factor of the disease is rarely taken into account. Molecular genetic studies make it possible to establish true cause and pathogenesis of the pathological process of hereditary diseases. Isolated congenital cataract is a group of diseases caused by the severe clinical polymorphism and genetic heterogeneity. These signs are the main reasons for the difficulties of diagnosing clinical-genetic form of the disease. According to literary data there are more than 50 genetic forms isolated hereditary cataracts. Clinical-diagnostic algorithm in patients with this pathology is complete only if there is medical genetic counseling of families by geneticists taking into account the received ophthalmological examinations and molecular genetic studies. Taking into account the verification of the etiological component of the pathological process, it becomes possible to plan correctly the preventive and curative measures. Currently, the main treatment for cataracts, is surgical. The functional state of the organ of vision, the prognosis of treatment, the stage of the pathological process and the somatic state of the patient determine the correction of therapeutic measures and in some cases contributes to the cancellation of surgical measures in cataracts. In such cases, the patient needs a conservative therapy. Currently, opacities of the lens are subjected to conservative curative schemes with the administration of drugs with metabolic effects in the form of eye drops.The pharmaceutical market is represented by a relatively small range of drugs of this group. Pirenoxine 75 mg in the form of eye drops (Catalin) is one of the known drugs used in ophthalmology practice in more than 12 countries. Catalin is applicable in the age-dependent cataract of mild or moderate degree of turbidity of the lens masses.
Keywords: cataract, hereditary diseases, medical genetics, clinical polymorphism, heterogeneity, prevention, treatment, pirenoxin.
For citation: Kadyshev V.V., Zinchenko R.A., Kutsev S.I. Clinical and genetic aspects of isolated hereditary cataracts // RMJ. Clinical ophthalmology. 2017. № 2. P. 114–118.
Освещены клинико-генетические аспекты наследственных изолированных катаракт
Введение
Одним из наиболее распространенных заболеваний органа зрения является катаракта, при диагностике которой необходимо учитывать этиологический фактор. В данной статье обсуждается наследственная катаракта, не являющаяся симптомом наследственного синдрома. Врожденная, ювенильная катаракта и катаракта зрелого возраста являются видами наследственных катаракт. Превалирующий объем инвалидности у детей с офтальмологическими заболеваниями приходится на врожденную катаракту, которая в структуре слепоты и слабовидения составляет 37% [8, 7]. Установлено, что 30–40% случаев заболевания обусловлены генетическими причинами [9].
Распространенность наследственных катаракт в различных популяциях РФ составляет в среднем 1,2:10 тыс. населения [5, 7]. Известно более 300 клинико-генетических вариантов данной нозологической формы [10]. Необходимо отметить, что структура клинико-генетических форм катаракты весьма разнообразна: наследственные изолированные катаракты (16–17%), катаракта вследствие наследственных нарушений обмена веществ и синдромальной моногенной патологии (74–75%), помутнения хрусталика, являющиеся симптомом хромосомной патологии (7–8%).
В практической деятельности врача-офтальмолога наследственные изолированные катаракты представлены 32 клиническими формами, каждая из которых характеризуется широким клиническим полиморфизмом. Врачами-генетиками с учетом этиологического компонента диагностировано более 50 генетических вариантов с изменениями в заинтересованных генах или локусах [3, 10].
По литературным данным, для наследственных катаракт описаны аутосомно-доминантный, аутосомно-рецессивный, Х-сцепленный рецессивный и митохондриальный типы наследования. Основная доля заболеваний (68%) ассоциирована с аутосомно-доминантным типом наследования [4].
Верификация формы патологии хрусталика значительно затруднена из-за выраженного клинического полиморфизма и генетической гетерогенности. Медико-генетическое консультирование, сбор подробного анамнеза, дополнительное обследование родственников (с целью выявления субклинического течения заболевания и уточнения типа наследования) необходимы для разработки профилактических мероприятий и применения новых методов терапии.
Виды наследственных изолированных катаракт
Все наследственные катаракты условно подразделяются по локализации патологического процесса и характеру помутнений хрусталиковых масс.
Полярная катаракта характеризуется помутнением округлой формы, локализованным на переднем и/или заднем полюсах капсулы хрусталика. Тип наследования – аутосомно-доминантный. Идентифицированы 4 гена: CRYAB, EPHA, CHMP4B, PITX3.
При ядерной катаракте патологический процесс локализован в ядре хрусталика. По форме помутнений выделяют пылевидную, зонулярную, полную катаракту. Данная подгруппа заболеваний является одной из самых генетически гетерогенных – диагностировано 12 генетических вариантов. Идентифицированы 6 заинтересованных генов: BFSP2, GJA8, CRYGD, CRYBB1, CRYBA1, CRYAA. По типу наследования выделяют аутосомно-доминантный и аутосомно-рецессивный варианты.
Зонулярная наследственная катаракта дебютирует в течение 1-го года жизни. При биомикроскопии (фокальном осмотре) помутнение имеет вид колец. При наведении световой щели сбоку катаракта имеет вид мутных слоев, расположенных в различных отделах хрусталика. Основной тип наследования – аутосомно-доминантный. Идентифицированы изменения в 5 генах: CRYBA4, CRYGC, MIP, HSF4, MAF.
Особенностью пленчатой катаракты является самопроизвольная резорбция хрусталиковых масс. В основном наследуется по аутосомно-доминантному типу.
Полная (тотальная) катаракта – помутнение всех слоев хрусталика. Однако биомикроскопически отмечается разная степень интенсивности патологического процесса. Тотальная катаракта в основном является двусторонней. Известны 2 типа наследования данных форм изменения хрусталиковых масс: аутосомно-доминантный и Х-сцепленный рецессивный.
Наиболее сложная в диагностическом плане форма наследственной катаракты – частичная атипичная, что связано с выраженным клиническим полиморфизмом. Помутнения могут быть различной формы, интенсивности, содержать кальцификаты. Наследуется по аутосомно-доминантному и аутосомно-рецессивному типу. В настоящее время выявлены изменения в генах GJA3, MIP, CRYGD.
При пылевидной наследственной катаракте помутнения представляют собой взвеси и локализуются в области ядра и кортикальном слое хрусталика. Данный вид катаракт характеризуется клиническим полиморфизмом (LIM2, CRYBB1, CRYGC).
В случае биомикроскопических находок изменений хрусталика в виде голубых точечных помутнений офтальмолог диагностирует лазурную катаракту. Тип наследования – аутосомно-доминантный (MAF, CRYBB2).
Особенности диагностики изолированных наследственных катаракт
Профилактика и лечение
Результаты комплексного обследования пациента позволяют сформировать план профилактических и лечебных мероприятий.
Различают первичную, вторичную и третичную профилактику наследственной патологии. Целью первичной профилактики является предупреждение рождения больного ребенка. Вторичная профилактика проводится на пренатальном уровне. При третичной профилактике наследственной патологии проводят коррекцию лечения с учетом клинической картины заболевания (на постнатальном уровне).
Особое внимание уделяется разработке новейших методов терапии катаракты и совершенствованию реабилитации пациентов [1, 2].
В настоящее время основным методом лечения катаракты является хирургический. При этом необходимо учитывать возраст манифестации заболевания, функциональное состояние сетчатки, интенсивность помутнения и возможность прогрессирования патологического процесса. В связи с этим оперативное лечение может быть отложено, и в таком случае необходимо проведение поддерживающей консервативной терапии. В настоящее время применяют препараты, действующие на метаболические процессы в хрусталике. В практической деятельности врача-офтальмолога используются препараты, содержащие пиреноксин, цитохром-С, аденозин, никотинамид, таурин. Пиреноксин 0,75 мг (Каталин) конкурентно ингибирует действие хиноновых веществ, продуцируемых в результате аномального метаболизма ароматических аминокислот, предотвращая развитие катаракты и ее прогрессирование. Каталин широко используется в 12 странах мира. В Российской Федерации глазная форма пиреноксина 0,75 мг успешно вернулась в арсенал врача в 2008 г. По данным отдаленного катамнеза, не менее чем у 47% пациентов отмечается положительная динамика заболевания. Препарат Каталин может быть рекомендован для консервативной поддерживающей терапии в случае помутнений хрусталика низкой или средней интенсивности в парацентральной области, когда хирургическое лечение отложено или не показано.
Исследование выполнено при частичной финансовой поддержке РНФ 17-15-01051

Наследственные заболевания передаются от одного или обоих родителей детям. Они вызываются генетическими мутациями, но далеко не все генетические заболевания являются наследственными. Как в этом разобраться, какие виды заболеваний бывают, как их лечить и как диагностировать — рассказываем в нашей статье.
Содержание
Что такое наследственные заболевания?
Наследственные заболевания — это заболевания, обусловленные генными или хромосомными мутациями. У людей от 20 000 до 25 000 генов. Генетическая мутация возникает, когда изменяется один или несколько генов. Если это генетическое изменение передается детям, то это наследственное генетическое заболевание.
При совпадении у партнеров статусов носительства определенных болезней есть высокий риск рождения ребенка с наследственным заболеванием. Если у вас не проявляются симптомы заболевания, вы по-прежнему можете быть носителем и передать мутации своим детям.
Многие генетически обусловленные заболевания проявляются не сразу после рождения, а спустя некоторое время. От наследственных заболеваний следует отличать врожденные заболевания, вызванные внутриутробными повреждениями, например, инфекцией или внешними воздействиями.
Чем отличаются наследственные заболевания от врожденных нарушений?
Генетические заболевания являются результатом изменения одного или нескольких генов и могут передаваться в поколениях или нет.
Все наследственные заболевания имеют генетическое происхождение, т. е. являются результатом изменения одного или нескольких генов и передаются из поколения в поколение. Симптомы могут не проявляться с самого рождения.
Врожденные нарушения могут быть наследственными или нет, а симптомы могут проявляться с рождения. Но их появление не обязательно связано с генетикой.
Виды наследственных заболеваний
Наследственные заболевания разделяются на хромосомные, генные и митохондриальные.
Хромосомные заболевания
В настоящее время описано около 1000 форм хромосомных заболеваний. Хромосомные заболевания возникают в результате изменения числа или структуры хромосом. Они характеризуются общими признаками: маленькая масса и длина тела при рождении, отставание в умственном и физическом развитии, задержка и аномалии полового развития и прочее.
Моногенные заболевания
Моногенные заболевания возникают в результате повреждения ДНК на уровне гена. Количество моногенных заболеваний по некоторым оценкам достигает 5000.
Среди признаков моногенных болезней можно выделить: различные формы умственной отсталости, дефекты органов слуха, зрения, скелетные дисплазии, болезни нервной, эндокринной, иммунной и других систем. К числу наиболее известных моногенных болезней относятся муковисцидоз, гемофилия А и В, болезнь Гоше, миодистрофия Дюшенна/Беккера, спинальная мышечная атрофия, дальтонизм.
Выявить тяжелые моногенные заболевания можно с помощью пренатальной диагностики, а также, определив наличие мутаций у родителей с помощью генетического теста.
Интереснее всего мне было узнать об особенностях метаболизма. Именно поэтому я выбрала Атлас: только тут есть достаточно объемный раздел на эту тему. Например, всю жизнь я борюсь с весом, мигренью, болями в шее и спине, анемией.
Митохондриальные заболевания
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами в функционировании митохондрий, которые приводят к нарушению тканевого дыхания.
Митохондрии содержат свою собственную ДНК. А болезни, вызванные мутациями в митохондриальной ДНК, наследуются исключительно по материнской линии. Если именно таким образом было унаследовано митохондриальное заболевание, существует 100% вероятность того, что каждый ребенок в семье его унаследует.
Симптомы могут включать в себя: нарушение роста, слабость мышц, аутизм, ментальные расстройства, проблемы с дыханием, слухом и зрением. Примеры митохондриальных заболеваний: синдром Лея, синдром Вольфа-Паркинсона-Уайта, наследственная оптическая нейропатия Лебера и другие.
Полигенные или мультифакториальные заболевания
Существуют также болезни с наследственной предрасположенностью, которые называют мультифакториальными или полигенными заболеваниями.
Мультифакториальные заболевания обусловлены наследственными факторами риска, и в значительной степени — неблагоприятным воздействием среды. К мультифакториальным заболеваниям относятся большинство хронических заболеваний, включая сердечно-сосудистые, эндокринные, иммунные, нервно-психические, онкологические и др. Например, бронхиальная астма, сахарный диабет, ревматоидный артрит, гипертоническая болезнь сердца и т.д.
Как передаются наследственные заболевания?
Организм человека состоит из триллионов клеток. Каждая клетка имеет ядро, которое содержит хромосомы. Каждая хромосома состоит из плотно свернутых нитей дезоксирибонуклеиновой кислоты (ДНК).
Гены — это инструкции по сборке белков в нашем организме, которые определяют специфические черты каждого человека, например, цвет глаз или волос. Большинство клеток в организме обычно содержат 46 хромосом, организованных в 23 пары. В каждой из этих 23 пар есть одна унаследованная хромосома от отца и одна — от матери. Из 23 пар 22 пары одинаковые у женских и мужских организмов, а одна оставшаяся определяет, являетесь вы мужчиной (XY) или женщиной (XX).
Мутации, из-за которых возникают наследственные заболевания, могут иметь доминантный или рецессивный характер наследования.
Доминантное наследование означает, что только одна копия гена — от матери или отца — должна иметь мутацию (или патогенный вариант гена) для проявления признака или заболевания. А при рецессивном типе человек наследует две измененные копии одного и того же гена.
Аутосомно-доминантный паттерн наследования
При аутосомно-доминантном наследовании заболеваний генетически обусловленная болезнь проявляется в том случае, если у человека есть хотя бы один мутированный ген, и этот ген не расположен на половых (Х и Y) хромосомах.
Болезнь Хантингтона и синдром Марфана — два примера аутосомно-доминантных болезней. Мутации в генах BRCA1 и BRCA2, которые также связаны с раком молочной железы, передаются по этой схеме.
Аутосомно-рецессивный паттерн наследования
При аутосомно-рецессивном наследовании мутируют обе копии генов. Чтобы унаследовать аутосомно — рецессивное заболевание, такое как муковисцидоз, спинальная мышечная атрофия, или фенилкетонурия (ФКУ), оба родителя должны быть носителями. Ребенок наследует две копии дефектного гена — по одной от каждого родителя. Например, люди, имеющие одну копию гена с мутацией, а вторую — без мутации, называются носителями, потому что сами они здоровы.
Х-сцепленное рецессивное наследование
В Х-сцепленном рецессивном наследовании мутированный ген находится на Х-хромосоме. Болезнь проявляется только в случае, если другой Х-хромосомы с нормальной копией того же гена у человека нет.
Мышечная дистрофия Дюшенна, некоторые виды дальтонизма и гемофилия А — примеры рецессивных заболеваний, связанных с X-хромосомой. Мужчина с рецессивным заболеванием, связанным с X-хромосомой, передаст свою нетронутую Y-хромосому сыновьям, и ни один из них не пострадает. Если он передаст свою Х-хромосому (с дефектным геном) своим дочерям, то все они будут носителями болезни. У его дочерей может не быть симптомов или только легкие признаки заболевания, но они могут передать мутированный ген своим детям.
Женщины-носители рецессивного заболевания, связанного с X-хромосомой, часто имеют лёгкие признаки заболевания или вообще не имеют симптомов. Это связано с тем, что у женщин-носителей есть одна нормальная копия гена и одна мутированная копия. Нормальная копия обычно компенсирует дефектную копию в женском организме, в отличие от мужчин, у которых только одна X-хромосома.
Женщины, имеющие только один патологический ген, передают заболевание в среднем половине своих детей вне зависимости от пола. Женщины же, имеющие два патологических гена, передают заболевание всем своим детям. К таким заболеваниям относятся гемофилия А и дальтонизм.

Как генетическое тестирование помогает при планировании семьи
Если вы знаете или предполагаете, что у вас или вашего партнера в семейной истории есть какое-либо генетическое заболевание, вы можете определить это с помощью Генетического теста Атлас. Генетическое консультирование поможет вам узнать о методах лечения, профилактических мерах и репродуктивных возможностях.
Как лечить наследственные заболевания и как с ними жить?
Раньше наследственные заболевания были неизлечимы. Сейчас это по-прежнему остаётся проблемой для многих заболеваний, но для некоторых из них методы лечения уже найдены. Например, это касается болезней, связанных с нарушением метаболизма.
При большинстве наследственных нарушений обмена веществ один фермент либо вообще не вырабатывается организмом, либо вырабатывается в форме, которая не работает. Например, при отсутствии какого-либо фермента в организме могут накапливаться токсичные вещества или может не синтезироваться необходимый продукт — как при гемохроматозе 1 типа.
При этом заболевании организм поглощает слишком много железа из пищи и не может естественным образом избавиться от избытка. Это может привести к чрезмерному накоплению железа в сердце, поджелудочной железе и печени.
Лечение генетических нарушений обмена веществ следует двум общим принципам:
- Необходимо сократить или исключить прием любой пищи или лекарств, которые не усваиваются организмом.
- Заменить или восполнить отсутствующий или неактивный фермент для восстановления метаболизма с помощью диеты и/или лекарств.
Есть более серьезные и распространенные наследственные заболевания, которые не лечатся. Например, мековисцидоз — скопление слизи в лёгких и в пищеварительной системе. От муковисцидоза нет лекарства, но разные методы контроля симптомов помогают предотвращать или уменьшать осложнения и облегчать жизнь с этим заболеванием.
Со временем муковисцидоз прогрессирует и может привести к летальному исходу, особенно при наличии сопутствующих инфекций. Сегодня благодаря достижениям медицины около половины людей с муковисцидозом доживают до 40 лет. Дети, рожденные с этим заболеванием в наши дни, смогут прожить ещё дольше.
Одно из самых тяжелых наследственных заболеваний, спинальная мышечная атрофия, также с недавнего времени поддается лечению с помощью генной терапии. Но доступен этот метод далеко не каждому. Препарат для лечения СМА — самый дорогой лекарственный препарат в мире.
Как я могу узнать, что являюсь носителем генетического заболевания?
Наши гены содержат инструкции, которые сообщают организму, как правильно функционировать. При изменении этих инструкций развиваются различные заболевания. Во многих случаях симптомы впервые проявляются в зрелом возрасте, поэтому иногда мы не знаем, что являемся носителями. Предупредить риски развития и передачи наследственного заболевания можно с помощью Генетического теста Атлас.

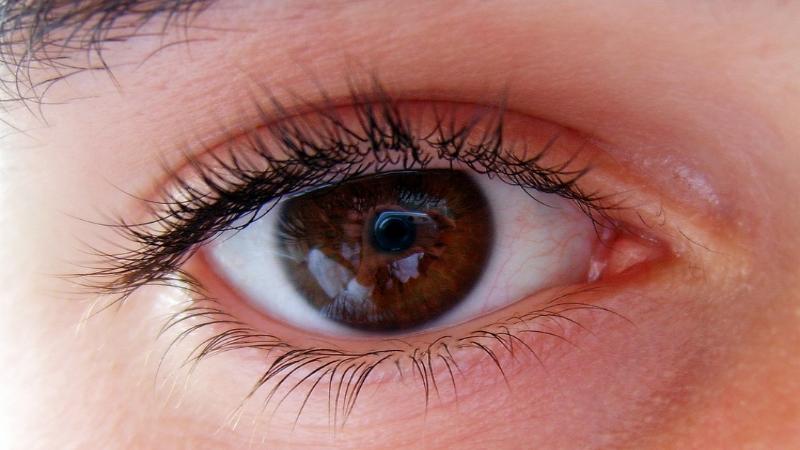
Сетчатка - это тонкая мембрана, расположенная на задней стороне нашего глаза. Именно эта чувствительная к свету нервная ткань контролирует то, как и что мы видим. Она содержит чувствительные к свету клетки фоторецепторов, которые преобразуют сфокусированное на сетчатке изображение в электрические импульсы, транслируемые с помощью зрительного нерва в наш мозг.
Заболевания сетчатки могут быть связаны с разными частями этого важнейшего элемента нашего глаза и привести к ухудшению зрения и даже слепоте. Болезни, влияющее на края сетчатки, влияют на наше периферийное зрение. Некоторые заболевания влияют на ту часть сетчатки, которая отвечает за центральное зрение.
Что такое наследственные дистрофии сетчатки?
Наследственные или генетические дистрофии сетчатки глаза - это болезни, вызванные мутациями наших ДНК. Эти нарушения в ДНК могут быть унаследованы от наших родителей или приобретены случайным образом. И когда ДНК, которая отвечает за нормальное функционирование сетчатки, повреждена, это может привести к невозможности воспроизведения важных белков, которые необходимы для нормального функционирования клеток сетчатки или наоборот - производить их в таком количестве, что это приносит больше вреда, чем пользы.
Виды заболеваний
Наследственные дегенеративные заболевания сетчатки - это целый ряд болезней, которые отличаются по своей патологии, симптомам, а также последствиями. И очень трудно предсказать, насколько сильно ухудшиться зрение и как быстро будет прогрессировать заболевание, но во многих случаях люди с наследственной дистрофией сетчатки могут полностью потерять свое зрение.
Ниже приведен список некоторых наследственных или генетических дистрофий сетчатки (список не полный, более того ряд болезней вообще не исследован):
Сотовая дистрофия сетчатки Doyne
Синий колбочковый монохроматизм
Самые часто встречающиеся виды дистрофии сетчатки - это возрастная макулярная дистрофия, которая в основном встречается у людей в возрасте 60 лет и старше, и пигментный ретинит, от которого в первую очередь страдают люди в возрасте от 20 до 60 лет. Во всем мире более 170 млн людей потеряли зрение из-за возрастной макулярной дистрофии, а пигментный ретинит стал причиной слабовидения или слепоты у 1.5 млн пациентов.
Некоторые из этих заболеваний хорошо известны и в связи с сопутствующим им болезням и симптомам. Например, синдром Ашера (Ушера) связан не только с ухудшением зрения, но и с глухотой, а синдром Барде-Бигля оказывает влияние и на другие части тела - он вызывает, в частности, задержку в развитии, ожирение и почечную недостаточность.
Характерные симптомы и проявления дистрофии сетчатки
Симптомы болезней сетчатки сильно отличаются и зависят от типа болезни. Но среди более общих симптомов, которые может замечать у себя человек, стоит упомянуть:
Внезапное резкое ухудшение зрения
Потеря периферийного зрения
Потеря центрального зрения
Световые вспышки в поле зрения
Изменение восприятия цветов
Потеря зрения в определенных областях зрения
Трудности с адаптацией к изменениям освещенности
Нистагм (непроизвольные колебательные движения глаз высокой частоты)
При появлении хотя бы одного из этих симптомов, рекомендуется немедленно обратиться к врачу-офтальмологу для диагностики и начала лечения.
Как диагностируются наследственные дистрофии сетчатки?
Самым важным первым шагом для диагностики наследственного заболевания сетчатки является изучение полной медицинской истории всей семьи пациента. Обычная проверка зрения может и не определить заболевание, несмотря на выраженные симптомы. Поэтому для точного диагностирования часто рекомендуют:
пройти процедуру оптической когерентной томографии (ОКТ) - исследование глазных тканей, при котором используется оптическое излучение ближнего инфракрасного диапазона, позволяющее дифференцировать патологические изменения сетчатки;
сделать фундус-фотографию, позволяющую получить морфологическое изображение глаза;
провести процедуру флуоресцентной ангиографии сетчатки (метод исследования сети сосудов и капилляров сетчатки, переднего отдела глазного дна и хориоидеи* с помощью их контрастирования флуоресцеином);
провести энтоптометрию (оценку функционального состояния сетчатки и всего зрительного анализатора);
провести тестирование поля зрения и цветового зрения,
В дальнейшем может понадобиться и генетическое тестирование, что позволит определить наличие дефектных генов, которые стали причиной заболевания. И это особенно очень важно, поскольку в настоящее время разрабатываются новые технологии лечения наследственных заболеваний глаз, которые основаны на оказании воздействия непосредственно на мутировавшие гены в сетчатке человека.
Кроме того, поскольку наследственная дистрофия сетчатки часто связана с нарушением деятельности и других органов тела, важно, чтобы врач предпринял меры по обнаружению и этих проблем, а также назначил лечение.
Два вида дистрофии сетчатки - болезнь Штаргардта и амавроз Лебера - достаточно часто неправильно диагностируются. Это связано с тем, что, хотя функция сетчатки у детей с этими заболеваниями нарушена, при стандартном исследовании зрения структурные нарушения могут быть и не замечены. Очень часто дети с такими расстройствами зрения вынуждены неоднократно обращаться к разным врачам, включая неврологов и офтальмологов, прежде чем их болезнь будет правильно диагностирована.
Для правильной диагностики болезни Штаргардта очень полезно тестирование остроты зрения и определение поля зрения. Кроме того, уменьшить число возможных вариантов диагноза может генетическое тестирование, поскольку многие дети с болезнью Штаргардта имеют поврежденный ген ABCA4.
У детей с амаврозом Лебера потеря зрения наступает рано и имеет самые тяжелые последствия. У таких детей очень часто также отмечается нистагм. Но, если не сделать электроретинограмму и генетический анализ, это заболевание можно легко спутать с окулокутанным альбинизмом**, который также отличается ухудшением зрения и нистагмом.
Лечение наследственных дистрофии сетчатки
За исключением возрастной макулярной дистрофии влажного типа, лечение которой может быть успешным, если она была вовремя диагностирована, большая часть наследственных дегенеративных заболеваний сетчатки в настоящее время, к сожалению, неизлечима. Специалисты по слабовидению могут подсказать как максимально использовать оставшийся потенциал зрения с помощью специальных очков или увеличительных стекол. Пациенты также должны регулярно проходить контрольное обследование у врача для того, чтобы убедиться, что у них не развиваются какие-либо побочные заболевания, связанные с болезнью сетчатки. Например, пациентам с болезнью Штаргардта, болезнью Беста или макулярной паттерн-дистрофией необходимо периодически проверяться в связи с возможным возникновением хориоидальной неоваскуляризации (врастание новообразованных сосудов под пигментный эпителий сетчатки). А пациентам с пигментным ретинитом необходимо проходить регулярное обследование в связи с возможной катарактой или появлением макулярного отека. Все эти осложнения в настоящее время лечат с помощью инъекций, глазных капель или операцией на катаракте.
Применяемая сегодня медикаментозная терапия предназначена в основном для замедления процессов, приводящих к ухудшению зрения, но не позволяет его восстановить.
Будущее лечения наследственных дистрофий сетчатки
Современные исследования дают нам основания надеяться, что ближайшее будущее даст нам наконец долгожданное лечение от наследственных заболеваний сетчатки глаз. Некоторые из идущих сейчас исследований концентрируются на генной терапии, которую сейчас считают наиболее перспективным методом будущего лечения наследственных форм дистрофии сетчатки. Этот метод заключается в замене дефектных генов или компенсации их действия, вызывающего заболевание. Например, уже были опубликованы очень впечатляющие результаты использования генной терапии для лечения амавроза Лебера, есть аналогичные результаты и в лечении пигментного ретинита.
За последние годы были идентифицированы большое количество генов, ответственных за возникновение дистрофии сетчатки. В настоящее время уже известны 242 типа различных генетических расстройств, связанных с дефектами в 202 разных генов. Например, мутации как минимум 23 генов вызывают появление аутосомно-доминантный пигментный ретинит с почти одинаковыми симптомами и протеканием болезни. В свою очередь, мутации гена ABCA4 могут вызвать возникновение болезни Штаргардта, а также колбочко-палочковой дистрофии или пигментного ретинита.
Сейчас также тестируется технология, в которой для лечения дегенеративных заболеваний глаз используются стволовые клетки. При этом лечении больным имплантируют клетки, например, пигментного эпителия сетчатки, которые были получены из стволовых клеток, что, по ожиданиям ученых, позволит не только замедлить развитие болезни, но и, возможно, восстановить зрение.
Еще одним интересным направлением являются бионические импланты. Уже сейчас одобрено применение импланта Argus II и идет разработка более совершенных моделей.
* Хориоидея — собственно сосудистая оболочка глаза. Хориоидея питает сетчатку и восстанавливает постоянно распадающиеся зрительные вещества. Она расположена под склерой. Капиллярное ложе хориоидеи опосредованно снабжает кислородом и питательными веществами фоторецепторы.
** Окулокутанный альбинизм - редкая группа наследственных заболеваний, выражающихся во врожденном отсутствии пигмента в коже, глазах и волосах.
Присоединиться


Справочник
Рекомендуем







Договор № 1
публичная оферта о добровольном пожертвовании
1. Общие положения о публичной оферте
2. Предмет договора
2.1. По настоящему договору Жертвователь в качестве добровольного пожертвования перечисляет собственные денежные средства на расчётный счёт Благополучателя, а Благополучатель принимает пожертвование и использует на уставные цели.
2.2. Выполнение Жертвователем действий по настоящему договору является пожертвованием в соответствии со статьей 582 Гражданского кодекса Российской Федерации.
3. Деятельность Благополучателя
4. Заключение договора
4.1. Акцептовать Оферту и тем самым заключить с Благополучателем Договор вправе физические и юридические лица или их представители.
4.2. Датой акцепта Оферты и соответственно датой заключения Договора является дата зачисления денежных средств на расчетный счёт Благополучателя либо, в соответствующих случаях, на счет Благополучателя в платежной системе. Местом заключения Договора считается город Москва Российской Федерации. В соответствии с пунктом 3 статьи 434 Гражданского кодекса Российской Федерации Договор считается заключенным в письменной форме.
4.3. Условия Договора определяются Офертой в редакции (с учётом изменений и дополнений), действующей (действующих) на день оформления платёжного распоряжения или день внесения им наличных денег в кассу Благополучателя.
5. Внесение пожертвования
6. Права и обязанности сторон
7. Прочие условия
7.1. В случае возникновения споров и разногласий между Сторонами по настоящему договору, они будут по возможности разрешаться путем переговоров. В случае невозможности разрешения спора путем переговоров, споры и разногласия могут решаться в соответствии с действующим законодательством Российской Федерации в судебных инстанциях по месту нахождения Благополучателя.
8. Реквизиты
Юр.адрес: 127422,г. Москва, Дмитровский проезд, дом 6, корпус 1,квартира 122,
ОГРН 1167700058283
ИНН 7713416237
КПП 771301001
Президент Байбарин К.А.
Наш сайт подключен к интернет-эквайрингу, и Вы можете внести пожертвование банковской картой Visa или Mastercard. После подтверждения выбранной суммы пожертвования, откроется защищенное окно с платежной страницей процессингового центра CloudPayments, где Вам необходимо ввести данные Вашей банковской карты. Для дополнительной аутентификации держателя карты используется протокол 3D Secure. Если Ваш Банк поддерживает данную технологию, Вы будете перенаправлены на его сервер для дополнительной идентификации. Информацию о правилах и методах дополнительной идентификации уточняйте в Банке, выдавшем Вам банковскую карту.
Гарантии безопасности
Процессинговый центр CloudPayments защищает и обрабатывает данные Вашей банковской карты по стандарту безопасности PCI DSS 3.0. Передача информации в платежный шлюз происходит с применением технологии шифрования SSL. Дальнейшая передача информации происходит по закрытым банковским сетям, имеющим наивысший уровень надежности. CloudPayments не передает данные Вашей карты нам и иным третьим лицам. Для дополнительной аутентификации держателя карты используется протокол 3D Secure.
Безопасность онлайн платежей
Предоставляемая Вами персональная информация (имя, адрес, телефон, e-mail, номер кредитной карты) является конфиденциальной и не подлежит разглашению. Данные Вашей кредитной карты передаются только в зашифрованном виде и не сохраняются на нашем Web-сервере.


2. Использование информации
Мы используем персонифицированную информацию конкретного посетителя сайта исключительно для обеспечения ему качественного оказания услуг и их учета. Мы не раскрываем персонифицированных данных одних посетителей сайта URL другим посетителям сайта. Мы никогда не публикуем персонифицированную информацию в открытом доступе и не передаем ее третьим лицам Исключением являются лишь ситуации, когда предоставление такой информации уполномоченным государственным органам предписано действующим законодательством Российской Федерации. Мы публикуем и распространяем только отчеты, построенные на основании собранных анонимных данных. При этом отчеты не содержат информацию, по которой было бы возможным идентифицировать персонифицированные данные пользователей услуг. Мы также используем анонимные данные для внутреннего анализа, целью которого является развитие продуктов и услуг URL
4. Ограничение ответственности
Мы делаем все возможное для соблюдения настоящей политики конфиденциальности, однако, мы не можем гарантировать сохранность информации в случае воздействия факторов находящихся вне нашего влияния, результатом действия которых станет раскрытие информации. Сайт www.looktosee.ru и вся размещенная на нем информация представлены по принципу "как есть” без каких-либо гарантий. Мы не несем ответственности за неблагоприятные последствия, а также за любые убытки, причиненные вследствие ограничения доступа к сайту URL или вследствие посещения сайта и использования размещенной на нем информации.
Юр.адрес: 127422, г. Москва, Дмитровский проезд, дом 6, корпус 1, квартира 122
Читайте также:
- Как оспорить постановление губернатора
- Именное накопительное свидетельство кбр как получить деньги
- Раскрыть содержание понятия место причинения вреда какой смысл вложил в него российский законодатель
- К какой правовой семье относится япония
- Предоставление сведений которые помогут инвестору решить заинтересует ли его проект является целью