
У человека тремы и нарушение прикуса наследуются сцепленно как аутосомно доминантные признаки
Обновлено: 19.05.2024
Врожденные заболевания являются одной из распространенных причин младенческой и детской смертности, развития хронических болезней у детей и инвалидизации.
Быстрый переход
Врожденные заболевания могут быть обусловлены генетическими мутациями (передающимися по наследству или спонтанными), инфекциями матери во время беременности (цитомегаловирус, ветряная оспа, краснуха), воздействием лекарств и химических веществ, загрязняющих воздух, воду или пищу. Причины множества врожденных дефектов до сих пор неизвестны.
Генетическими или наследственными факторами обусловлены около 20 % врожденных заболеваний. К ним относятся нарушения, при которых мутация затрагивает один ген (серповидно-клеточная анемия); хромосомные нарушения, при которых хромосомы (или их части) отсутствуют (синдром Тернера) или имеют структурные изменения (увеличение количества хромосом или трисомия при синдроме Дауна); многофакторные нарушения, вызванные мутациями двух и более генов. Врожденные дефекты и нарушения развития могут вызывать делеции или дупликации отдельных генов (изменения митохондриальной ДНК). Примером такого заболевания является муковисцидоз, характеризующееся поражением экзокринных желез и жизненно-важных органов (легких и желудочно-кишечного тракта). Врожденные заболевания также ассоциированы со случайными (новыми) повреждениями генов, спонтанными (не наследующимися от родителей) мутациями (большинство случаев ахондроплазии).
По статистике, врожденные заболевания имеют 2–3 % младенцев. К возрасту 1 года их число увеличивается до 5 %, поскольку не все эти патологии диагностируются сразу после рождения.
Врожденные (наследственные) заболевания также классифицируют по типу наследования. При аутосомно-доминантном наследовании заболевание может передаваться от родителя к ребенку в 50 % случаев (мышечная дистрофия Дюшенна, хорея Гентингтона). При аутосомно-рецессивном наследовании генетическая аномалия передается ребенку только в том случае, если оба родителя наделены одним и тем же дефектным геном (муковисцидоз, серповидно-клеточная анемия). Здесь частота наследования составит 25 %, то есть в среднем у 1 ребенка из 4 детей этих родителей будет аутосомно-рецессивное заболевание.
Патологические состояния могут передаваться и при наследовании, сцепленном с полом (наследование гена, находящегося в половых хромосомах). Х-сцепленные рецессивные заболевания почти всегда ограничены мужским полом (гемофилия, дальтонизм, мышечная дистрофия Дюшенна). Х-сцепленные доминантные заболевания встречаются как у мальчиков, так и у девочек, однако у детей мужского пола имеют более тяжелое течение (Х-сцепленный гипофосфатемический рахит). Y-сцепленные заболевания встречаются довольно редко, поскольку Y-хромосома содержит всего несколько генов (ихтиоз).
Некоторые врожденные заболевания формально не относятся к генетическим, но имеют ту или иную выраженность наследования: наследуются факторы риска либо сам ген, но с низкой пенетрантностью (частотой проявления гена в признаках).
Гемофилия
Гемофилия — группа редких наследственных нарушений свертываемости крови, вызванных дефицитом необходимого белка (фактора свертывания крови).
Гемофилия A (классическая) встречается чаще (>80 % случаев) и связана с дефицитом VIII фактора свертывания крови, гемофилия B (болезнь Кристмаса) — реже (>10 % случаев), она обусловлена недостаточностью IX фактора свертывания крови. Гемофилия C встречается очень редко, обусловлена дефицитом XI фактора свертывания крови, чаще всего в классификации группы ее не упоминают.
Заболевание относится к X-сцепленным с полом, наследуется по рецессивному признаку по женской линии. Классическую гемофилию вызывают мутации гена F8, расположенного на X-хромосоме. Примерно в 70 % случаев заболевание наследуется по Х-сцепленному образцу, в остальных случаях оно возникает спонтанно (новая мутация), в дальнейшем спонтанное заболевание становится наследственным. Гемофилией A болеют практически исключительно мужчины, редкие случаи заболевания у женщин, носительниц дефектного гена, почти всегда характеризуются легким течением. Гемофилию B вызывают мутации гена F9, так же расположенного на X-хромосоме, она характерна для мужчин, женщины болеют очень редко. В некоторых случаях заболевание возникает спонтанно (приобретенная гемофилия A или B) и тоже связано с недостаточностью VIII или IX факторов свертывания крови. Приобретенная гемофилия является аутоиммунным заболеванием, при котором организм вырабатывает антитела, атакующие факторы свертывания крови (чаще всего VIII фактор). Примерно в половине случаев приобретенной гемофилии у пациента имеется связанное с ней основное состояние или заболевание (беременность, аутоиммунные заболевания, миелопролиферативные заболевания, воспалительные заболевания кишечника и др.), в остальных эпизодах причина остается невыясненной.
Гемофилия A затрагивает примерно 1 из 5 000 новорожденных мальчиков, гемофилия B — примерно 1 из 25 000. Около 60 % пациентов имеют тяжелую форму гемофилии, обычно им ставят диагноз при рождении или в течение первых 2 лет жизни.
Возраст манифестации гемофилии и тяжесть течения заболевания зависят от уровня активности факторов свертывания крови. Легкая форма характеризуется уровнем фактора свертывания крови (VIII или IX), превышающим 5 % от нормы, средняя — 1–5 %, тяжелая — ниже 1 %. У большинства пациентов, независимо от тяжести течения заболевания, эпизоды кровотечения чаще встречаются в раннем детском, детском и подростковом возрасте, чем в дальнейшей взрослой жизни.
При легкой и умеренной формах гемофилии длительные кровотечения могут возникнуть только в результате травмы, хирургического вмешательства или стоматологической процедуры. Нередко диагноз гемофилии ребенку устанавливают к 5–6 годам, обратив внимание на длительное посттравматическое кровотечение, длительное кровотечение во время стоматологического лечения или операции. К другим заметным симптомам легкого и умеренного течения гемофилии относятся непроходящие гематомы (синяки), частые носовые кровотечения, кровоточивость десен.
Для тяжелой формы гемофилии характерны эпизоды спонтанного кровотечения, которые приводят к кровоизлияниям различной локализации — в мягкие ткани, мышцы, суставы. При гемартрозах (кровоизлияние в полость сустава) возникает ограничение подвижности суставов, сопровождающееся острой болью и воспалением. У пациентов с тяжелой формой гемофилии спонтанные кровотечения чаще всего происходят в мышцы и суставы, однако могут затрагивать любой внутренний орган, включая почки, органы ЖКТ, головной мозг (гематурия, мелена, гематохезия, желудочно-кишечные кровотечения, внутричерепные кровотечения). Тяжелая форма гемофилии A обычно проявляется в раннем детском возрасте, диагноз чаще всего устанавливается к 2 годам ребенка. Обычными симптомами (при отсутствии лечения заболевания) здесь являются кровотечения из-за незначительных травм ротовой полости (прикусывание губ, языка, щек), подкожные гематомы, большие шишки после удара головой, спонтанные кровотечения (2–5 эпизодов в месяц).
Диагноз гемофилии A или B устанавливается на основании симптомов, истории личного и семейного анамнеза пациента, лабораторных исследований (общего анализа крови, коагулограммы, оценивающей состояние системы свертывания крови и активности ее белков, молекулярно-генетического исследования). Парам из группы риска по рождению ребенка с гемофилией необходимо получить генетическую консультацию на этапе планирования беременности. Определение конкретной мутации гена F8 (или F9) помогает не только выявить женщин-носительниц дефектного гена в конкретной семье, но и полезно для пренатальной диагностики гемофилии (амниоцентез, биопсия хориона).
Некоторым пациентам с легкой формой гемофилии назначается синтетический аналог вазопрессина — десмопрессин, повышающий VIII фактор свертывания крови (вводится внутривенно или интраназально), а также антифибринолитические средства, замедляющие распад факторов свертывания крови.
Примерно в 30 % случаев лечения тяжелых случаев гемофилии длительная заместитетельная терапия может приводить к изоиммунизации — образованию антител к вводимым факторам свертывания крови (иммунная система распознает вводимый фактор VIII как чужеродный). Этот процесс может сопровождаться аллергическими реакциями (разной степени тяжести), возрастает риск жизнеугрожающих кровотечений. В этом случае пациенту назначается альтернативное лечение — плазмаферез, иммунодепрессанты.
Гемохроматозы
Гемохроматоз — наследственное заболевание, характеризующееся нарушением обмена железа и его накоплением в тканях и органах.
Заболевание сопровождается повышенным всасыванием железа в желудочно-кишечном тракте и накоплением в печени, сердце, поджелудочной железе, суставах, гипофизе, что приводит к полиорганной недостаточности и развитию таких болезней, как цирроз печени, рак печени, диабет, болезни сердца и суставов.
Наследственный гемохроматоз вызывают мутации генов HFE, HFE2, HAMP, SLC40A1 и TfR2, он классифицируется в зависимости от возраста начала, генетической причины и способа наследования: гемохроматоз 1 типа, гемохроматоз 2 типа, гемохроматоз 3 типа, гемохроматоз 4 типа. Вторичный гемохроматоз ассоциирован с другими заболеваниями (не является наследственным) — анемией, хронической болезнью печени, инфекциями.
Гемохроматоз, редко возникающий в младенческом возрасте и не имеющий явной причины, называют неонатальным гемохроматозом. При этой форме заболевания избыточное накопление железа в тканях и органах начинается еще до рождения ребенка. Неонатальный гемохроматоз быстро прогрессирует и характеризуется поражениями печени, которые выявляют при рождении или в первые дни жизни. Дети с этим заболеванием часто рождаются недоношенными или имеют нарушения внутриутробного развития. Точная причина неонатального гемохроматоза неизвестна, есть версия, что он развивается в том случае, если иммунная система матери распознает клетки печени ребенка как чужеродные. Симптомы обычно включают гипогликемию, нарушения свертываемости крови, пожелтение кожных покровов и склер глаз, отеки. Диагноз устанавливается на основании признаков и симптомов, лабораторных и инструментальных исследований, биопсии печени. Лечение включает переливание крови, внутривенное введение иммуноглобулинов, трансплантацию печени.
Гемохроматоз 1 типа ассоциирован с мутациями в гене HFE, расположенном на коротком плече 6–й хромосомы, наследуется по аутосомно-рецессивному типу, является наиболее распространенным типом наследственного гемохроматоза и поражает в основном мужчин. Вероятность того, что ребенок унаследует мутацию в гене HFE от родителя составляет 25 %, вероятность того, что он станет носителем дефектного гена — 50 %.
Начальные симптомы заболевания обычно отмечаются в возрасте 40–60 лет и включают боль в животе, снижение полового влечения, усталость, слабость, боли в суставах, сухость кожи. В дальнейшем проявляются такие симптомы и осложнения, как изменение пигментации кожи (бронзовая кожа), выпадение волос на голове и туловище, аритмия, кардиомиопатия, хроническая сердечная недостаточность, сахарный диабет, гепатомегалия, цирроз печени, спленомегалия, атрофия яичек, аменорея (у женщин) и другие.
Диагноз устанавливается на основании симптомов заболевания, лабораторных исследований (уровень железа, ферритина, трансферрина, железосвязывающей способности сыворотки крови, маркеры функции печени, уровень глюкозы крови и др.), инструментальных исследований (рентгенография суставов, электрокардиография и эхокардиография сердца, УЗИ органов брюшной полости, МРТ печени и др.), биопсии печени, молекулярно-генетического исследования (в том числе для выявления родственников — имеющих гемохроматоз или носителей).
Лечение гемохроматоза направлено на удаление избытка железа из организма (флеботомия, хелатирование) с помощью забора крови (как при взятии анализов или донорстве крови, только в большем объеме) и специальных препаратов, образующих комплексное соединение с железом и способствующих его удалению из железосодержащих белков (дефероксамин).
Терапевтическая флеботомия сначала проводится 1–2 раза в неделю, поддерживающая — каждые 2–4 месяца. Вместе с этим проводится симптоматическое лечение сахарного диабета, патологий сердца, цирроза печени и других заболеваний, вызванных гемохроматозом. Пациентам запрещен прием препаратов железа и любых медикаментозных средств или комплексов, которые могут содержать железо и витамин C, запрещен алкоголь во избежание дальнейшего повреждения печени (если оно имеется), рекомендуется придерживаться диеты, исключающей продукты с высоким содержанием железа (красное мясо, яблоки, печень, шпинат).
Гемохроматоз 2 типа вызывают мутации в генах HFE2 или HAMP, он наследуется по аутосомно-рецессивному типу и проявляется чаще всего в детском возрасте. Для этого типа заболевания характерны следующие симптомы: высокие уровни ферритина и трансферрина, врожденный фиброз печени, артропатия, кардиомиопатия, генерализованная гиперпигментация кожи, мышечная слабость, гипогонадизм, задержка полового созревания, сахарный диабет, цирроз печени, остеопороз, спленомегалия, гепатомегалия и другие. Диагностика и лечение гемохроматоза 2 типа проводятся аналогично диагностике и лечению гемохроматоза 1 типа.
Гемохроматоз 3 типа вызывают мутации в гене TFR2, он наследуется по аутосомно-рецессивному типу. Симптомы обычно проявляются до 30 лет и, помимо вышеперечисленных, могут включать снижение уровня лимфоцитов и нейтрофилов в крови, красные или пурпурные пятна на коже. Диагностика и лечение гемохроматоза 3 типа проводятся аналогично диагностике и лечению гемохроматоза 1 и 2 типов.
Симптомы гемохроматоза 4 типа могут проявиться как в детском, так и во взрослом возрасте. Этот тип заболевания вызывают мутации гена SLC40A1, наследуется оно по аутосомно-доминатному типу. Вероятность того, что ребенок унаследует мутацию в гене SLC40A1 от родителя составляет 50 %, соответственно, вероятность того, что он станет носителем дефектного гена — тоже 50 %. Симптомы гемохроматоза 4 типа могут проявляться в виде повышенной утомляемости, слабости, болей в суставах, изменения пигментации кожи, затруднения дыхания, сердечной недостаточности, сахарного диабета, анемии, врожденного фиброза печени, цирроза печени, остеоартроза, катаракты. Диагностика и лечение гемохроматоза 4 типа проводятся аналогично диагностике и лечению гемохроматоза 1, 2 и 3 типов.
Наследственный гемохроматоз — заболевание с пониженной пенетрантностью (вероятностью того, что ген будет иметь любые фенотипические проявления) дефектного гена, т. е. у некоторых людей с мутациями гена HFE никогда не проявятся симптомы, при этом у их детей или других членов семьи с мутацией гена произойдет манифестация заболевания.
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна — редкое наследственное заболевание, характеризующееся нарастающей мышечной слабостью с последующей атрофией мышц. Ассоциировано с мутацией гена DMD, расположенного на половой X-хромосоме (X-сцепленный рецессивный тип наследования).
Типы распределения моногенных заболеваний в родословных зависят в основном от двух показателей:
• является ли фенотип доминантным (проявляющимся, когда только одна хромосома из пары несет мутантный аллель, а другая хромосома имеет в этом локусе дикий тип аллеля) или рецессивным (проявляющимся, только когда обе хромосомы пары несут мутантные аллели в данном локусе);
• позиция гена на хромосоме, который может располагаться либо на аутосоме (хромосомы с 1 по 22), либо в половой хромосоме (Х- и Y-хромосомы).
Необходимо, тем не менее, различать гены, которые физически расположены в половых хромосомах (Х- или Y-синтения), и гены, которые наследуются сцепленно с Х- (или Y-) хромосомой. Большинство локусов на Х-хромосоме наследуются Х-сцепленно, поскольку они участвуют в мейотической рекомбинации только в женском гаметогенезе, когда есть две Х-хромосомы, и не могут рекомбинировать с Y-хромосомой в процессе мужского гаметогенеза.
Тем не менее есть несколько генов (названные псевдоаутосомными локусами, обсуждаемые далее в этой главе), располагающиеся в Х-хромосоме, но не проявляющие Х-сцепленного наследования, поскольку они могут рекомбинировать с аналогами в Y-хромосоме. Таким образом, выделяют четыре основных типа моногенного наследования (если объединить аутосомные и псевдоаутосомные типы вместе).
Кроме классических образцов родословных, наблюдаемых при патологии аллелей, расположенных в ядерных хромосомах, существует другой класс нарушений с отчетливым материнским типом наследования вследствие мутаций в митохондриальном геноме.
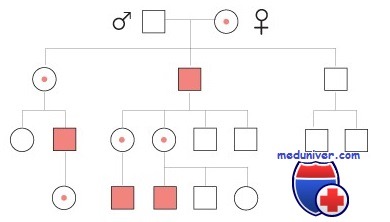
Х-сцепленное наследование
Аутосомное и Х-сцепленное наследование
Тот факт, находится ли аномальный ген в аутосоме или сцеплен с Х-хромосомой, имеет выраженное влияние на клиническое проявление болезни. Во-первых, аутосомные заболевания в основном влияют на мужчин и женщин одинаково.
Для Х-сцепленных болезней ситуация совсем другая. Мужчины имеют единственную Х-хромосому и, следовательно, гемизиготны по генам Х-хромосомы; мужчины 46,XY никогда не гетерозиготны по аллелям в локусах, расположенных на Х-хромосоме, тогда как женщины могут быть гетерозиготными или гомозиготными по данным локусам.
Во-вторых, чтобы компенсировать двойное количество генов Х-хромосомы у женщин, в каждой отдельной клетке аллели большинства Х-сцепленных генов экспрессируются только на одной из двух Х-хромосом.
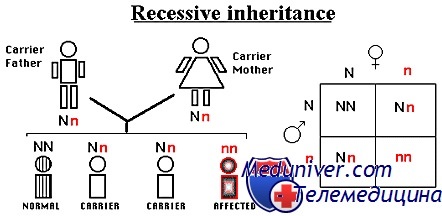
Рецессивное наследование
Рецессивное наследование
Классически считают, что фенотип, проявляющийся только у гомозигот (или, для признаков Х-сцепленных, у гемизиготных мужчин), но не у гетерозигот, — рецессивный. Большинство изученных к настоящему времени рецессивных заболеваний — следствие мутаций, снижающих или отключающих функцию продукта гена, так называемые мутации утраты функции.
Например, множество рецессивных болезней вызваны мутациями, которые нарушают функции ферментов. Они обычно наследуются как рецессивные болезни, поскольку гетерозиготы с функционирующим одним из пары аллелей производят достаточно продукта для нормальной физиологической функции (50% количества у гомозигот дикого типа), тем самым предотвращая болезнь.
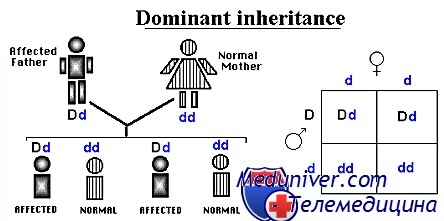
Доминантное наследование
Доминантное наследование
В отличие от этого, фенотип, проявляющийся как у гомозигот, так и у гетерозигот для мутантного аллеля, наследуется доминантно. Доминантное заболевание развивается независимо от наличия нормального продукта гена, производимого благодаря существующему нормальному аллелю. При чисто доминантной болезни гомозиготы и гетерозиготы по мутантному аллелю поражены одинаково.
Чисто доминантные заболевания редки, если вообще существуют в медицинской генетике. Иногда одновременно фенотипически проявляются два различных аллеля в локусе, в этом случае такие два аллеля называют кодоминантными. Известный пример кодоминантной экспрессии — система групп крови АВО. Чаще всего доминантное заболевание более тяжелое у гомозигот, чем у гетерозигот, в этом случае болезнь называют неполно доминантной (или полудоминантной). Различные молекулярные механизмы, объясняющие, почему определенные мутации вызывают доминантно, а не рецессивно наследуемые болезни.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021

Полидактилия (многопалость): признаки, лечение и ген полидактилии.
Полидактилия (многопалость, гипердактилия) — это заболевание, характерным признаком которого является наличие лишних пальцев на нижних либо верхних конечностях. Врождённая патология наблюдается у людей, кошек, собак и лошадей. Полидактилия у человека является распространённым наследственным заболеванием конечностей.
Ген полидактилии
Многопалость включена в список 120 генетических недугов. Если у одного человека доминирует ген полидактилии над нормальным строением конечности, а другой человек имеет только нормальный ген, то вероятность рождения ребёнка с многопалостью у этой пары составляет 50%. Многопалость имеет доминантный признак, а не рецессивный. Это значит, что полидактилия или шестипалость у человека определяется доминантным геном. Как доминантные признаки передаются, к примеру, не только полидактилия или многопалость, но и отсутствие малых коренных зубов.
Полидактилия наследуется по аутосомно-доминантному типу с определённым процентным соотношением. Саму мутацию определяет генная или геномная полидактилия.
Причины полидактилии
Исходя из МКБ (международной классификации болезней) полидактилия представляет собой анатомическую аномалию. Досконально о всех причинах появления заболевания не известно даже врачам. Причины многопалости: генетическая предрасположенность, хромосомные болезни, мутации на генном уровне. Особенность недуга состоит в наследовании аномалии. Болезнь полидактилия наследуется по аутосомно-доминантному типу с малой пенетрантностью (процентным соотношением). Это означает то, что наследуемый ген может быть задействован у одного из родителей, хотя он сам может не иметь данного заболевания. Зачастую недуг считается спровоцированным серьёзным хромосомным нарушением.
Полидактилия имеет виды в зависимости от формы удвоения:
1 тип — лишний отросток состоит только из мягких тканей;
2 тип — нарушение проявляется из-за раздвоения основного пальца;
3 тип — лишний палец считается сформированным и полноценным.
Классификация полидактилии: преаксикальная, постаксиальная, центральная.
Преаксикальная полидактилия — это дефект со стороны большого пальца. Постаксиальная полидактилия — это дефект со стороны мизинца.
Признаки полидактилии
Первые признаки можно обнаружить уже у новорождённого ребенка в виде добавочных пальцев. Полидактилия — это генная мутация, какая передаётся по наследству. Лишние пальцы могут быть представлены как в виде мягких отростков, так и иметь полноценную костную структуру. Мягкотканное образование является отростком без признаков функционирования. Пальцы могут иметь одинаковый размер с нормальными или же отличаться от них. Чаще всего, дополнительные отростки обладают меньшим размером.
Ещё одним признаком патологии является нарушение со стороны костно-суставной системы отростков. Наряду возможны аномальные нарушения грудной клетки, отклонения сердца, низкий рост. По мере взросления данная проблема может усугубиться. Люди, имеющие такое заболевание, зачастую вынуждены подбирать себе ортопедическую обувь.
Интересный факт! Синдром Лоуренса-Барде-Муна-Бидля сочетает в себе многопалость и отклонение в развитии. Присутствует большой лишний вес, деформация черепа, недоразвитость со стороны половых органов.
Полидактилия у детей
Полидактилия или шестипалость у человека — это пример наличия доминантного гена, носителем которого был один из родителей. Статистика показывает, что синдром полидактилия встречается у 1 ребёнка из 5000 родившихся детей. Многопалость является относительно редким заболеванием. С одной стороны кажется, что помимо эстетической составляющей этот недуг не несёт в себе негативных последствий. Патология оказывает отрицательное воздействие на качество жизни ребёнка и на его психологическое состояние, вплоть до ограничения в выборе специальности. Зачастую болезнь провоцирует отклонение в развитии конечностей и проблемы в функционировании опорно-двигательной системы.
Полидактилия у детей чаще всего наблюдается на руках. На ногах данная патология бывает немного реже. Однако, есть случаи, когда болезнь проявляется и так, и так. Недуг начинает формироваться в момент внутриутробного развития ребёнка, однако полидактилия может исчезнуть к 32 неделям.
Принимать решение об операции или отказаться от неё — дело индивидуальное. Однако, если наличие лишних пальцев мешает нормальному функционированию, то рекомендуется оперативное вмешательство, чтобы избежать отсталости в развитии. Болезнь оказывает негативное влияние на моторику рук и функциональную составляющую ног.
Если лишние пальцы не мешают при ходьбе и не препятствуют удобному ношению обуви, то операцию можно отметить или отложить. По достижению ребёнка взрослого возраста, он сам решит, необходима ли для него операция в косметических целях или нет. Необходимо отметить, что у 90% детей с наличием недуга нет прочих заболеваний.
Интересный факт! Если тест выявил отсутствие генетической наследственности заболевания, то болезнь может быть спровоцирована негативными факторами на 6-8 неделе беременности. К негативным факторам относятся: инфекции; вредные привычки; запрещённые при беременности лекарства; облучение.
Негативные причины могут оказать отрицательное влияние на формирование костной системы ребенка и развитию недуга. Для правильного формирования костного скелета и избегания болезни необходимо придерживаться здоровому образу жизни и исключить негативные факторы.
Полидактилия кисти
Полидактилия кисти характеризуется увеличением числа фаланг и трубчатых костей. Этот недуг наблюдается у людей чаще, чем аномалия стоп. Делится по мере локализации на: центральную (2-4 пальцев), радиальную (удвоение сегментов первого луча), ульнарную (пятого пальца). Патология может стать причиной порока развития или наследственных заболеваний.
Полидактилия стоп
Полидактилия стопы характеризуется наличием добавочных пальцев на ногах. Несмотря на то, что болезнь чаще всего затрагивает руки, есть меньший процент детей с полидактилией ног. Ещё реже можно встретить случаи смешанной формы болезни, где аномалия проявила себя как на верхних конечностях, так и на нижних.
Диагностика полидактилии
Для первичной диагностики необходим осмотр врача. Обычно новорождённых осматривают в роддоме. В осмотре ребёнка принимают участие несколько врачей, включая генетика, педиатра и ортопеда. Если в сведении об истории болезни присутствуют наследственные заболевания, то такой ребенок исследуется наиболее тщательным образом.
Для определения вида заболевания производится ряд обследований, включая рентгенологическое и генетическое. Для определения состояния мягких тканей проводится магнитно-резонансная томография. Полидактилия, имеющая аутосомно-доминантный тип наследования, может обнаружиться уже в первом триместре протекания беременности с помощью УЗИ. Тщательность выявления будет завесить от профессиональных навыков врача-узиста и от расположения плода.
Лечение полидактилии
Лечение полидактилии во всех случая носит исключительно оперативный характер. Если у младенца имеется несформированный палец из мягкой ткани, то его можно без проблем удалить в ближайший месяц. Однако, если палец полностью сформирован и функционален, оперировать ребёнка можно спустя год после рождения. Годовалый малыш считается оптимальным пациентом для такого вида хирургии и безопасного удаления. В таком возрасте кости ребёнка считаются достаточно сформированными, а здоровье стабильным. Оперирование назначается человеку после полной диагностики. Преаксикальную полидактилию оперируют методом удаления пальца, а вместе с ним всех связок, костей и сухожилий. При центральной полидактилии используется наиболее сложный вариант оперирования, может потребоваться даже не одна операция и реконструкция для того, чтобы полностью восстановить функциональность кисти. При постаксикальной полидактилии соединённый мягкой перегородкой с кистью палец удаляется в раннем возрасте с помощью однократной операции. При хорошо сформированном дополнительном пальце производится ряд операций по реконструкции мягких тканей, сухожилий и костей.
В послеоперационный период пациенту назначается комплекс лечебной физкультуры, физиопроцедуры (магнит, инфракрасное облучение), сеансы массажа. Если операция произведена вовремя, то неблагоприятный прогноз маловероятен. Период реабилитации зависит от индивидуальных особенностей организма, заживления и способа хирургического вмешательства. В запущенных случаях, после оперативных вмешательств, на восстановление может понадобиться несколько лет.
Вывод
Если на УЗИ у ребёнка обнаружили полидактилию, то это не повод для сильного беспокойства и паники. Полидактилия, что значит наличие гена, отвечающего за присутствие лишнего пальца или дополнительного отростка, без проблем оперируется без серьезных последствий. Современная медицина предоставляет возможность безопасного оперативного вмешательства по удалению аномалии. Однако, стоит понимать, что если у одного из родителей всё же была болезнь полидактилия до оперативного вмешательства, а ребенок родился без неё, то он всё равно является носителем. Полидактилия — это не приговор, а лишь нестандартное строение конечности.
Читайте также: